потенциометрия статьи
Возможно вас заинтересуют материалы со следующими тегами, по теме “потенциометрия”
Околотематические теги присвоенные страницам с тегом “потенциометрия”КАТЕГОРИИ
- Лабораторные приборы, оборудование
- Химия
- красители, хим. соединения
- химические соединения
- Удобрения
- растворы для лабораторных исследований
- противоморозные добавки
- техническая химия
- эпоксидные смолы
- Инструкции, руководства по эксплуатации
- Удобрения ГОСТы
- нефтехимия
- водорастворимые комплексные минеральные
- химическая продукция
- рН-метры, pH-метрия, измерение водородного показателя pH
- весовое оборудование
- водоочистка, приборы для очистки воды
- Госты, стандарты
- растворы для микробиологии, микробиология
- гальванические добавки
- органические растворители
- клеи
- ПАВ, Поверхностно-активные вещества
- индикаторная бумага
- отвердители, ускорители отверждения
- реагенты для водоподготовки
ТЕГИ
Электроды используемые с промышленными приборами
- 20 января 2019
- потенциометрия вода аналитическая химия электроды индикаторные электроды pH электрод титрование pH-электрод измерение ph ионы водорода промышленные приборы
Таблица электродов, используемых с промышленными приборами 1
|
Тип прибора |
Электроды, используемые с приборами / Electrodesusedwithdevices | |||
| ионоселективный | ед.изм. | редокс-метрический | электрод сравнения | |
| АН-7529М АН-7560М | 5М2.840.019 | рН | 5М2.840.072 | |
| АС-7932М | 5М2.840.074 | рН | 5М2.840.072 | |
| П-210 П-215И П-215М | Монтируются в арматуре ДПг-4М, ДМ-5М, используемой с преобразователем | ЭХСВ-1 (в системе выносного проточного электрода)* ЭВП-08 (погружнойнепроточный) / | ||
| ЭА-2 ЭМ-I-01 ЭМ-CN-01 ЭМ-Cl-01 ЭМ-NO 3 -07 ЭС-10-07 ЭСП-01-14 ЭСП-04-14 ЭСП-31-06 ЭСС-01 | pS pJ pCN pCl pNO 3 pNa рН рН рН pAg | ЭО-01 ЭПВ-1 ЭТП-02 | ||
| pNa-205 | ЭС-10-07 ЭСЛ-63-07СР ЭСЛ-43-07СР | pNa рН рН | ЭХСВ-1 ЭВП-08 | |
| СХ-2 | ЭЗ-01 ЭО-01 | ЭХСВ-1 ЭВП-8 ЭСП-04-14 (10) | ||
| СЦ-2 | ЭМ-CN-01 | рCN | ЭЗ-01 ЭА-2 | ЭХСВ-1 ЭВП-08 ЭСП-04-14 (4) |
* - при избыточном давлении рабочей среды от 0,2 до 0,6 МПа должен использоваться с регулятором давления (например, РДС-1)
Категории электродов: «Ион-селективные электроды»; «pH-электроды»; «датчики ОВП редокс-электроды»; «датчики проводимости»; «электроды вспомогательные (сравнения)»; «датчики температуры»; «кислородные датчики» по назначению: «электроды лабораторные»; «электроды промышленные»; «электроды промышленно-лабораторные»; «электроды ионоселективные»; «электроды вспомогательные»; по областям применения: «пищевые продукты и сырьё»; «полевые измерения»; «сильные кислоты и основания»; «измерения в малых объёмах»; «общелабораторные»; «промышленные»; Фильтр характеристик: «HANNA электроды»; «HANNA edge»; «с микрочипом»; «с температурным датчиком»; «с Bluetooth беспроводные электроды»; рабочий диапазон рН: 2 — 10 рН; 4 — 7 рН; 7 — 9 рН; 4,5 — 9,5; 0 — 11 рН; 5 — 7,5 рН; 1 — 10 рН; 5,5 — 8,5 рН; 0 — 8,5 рН; 2 — 8,5 рН; 1 — 9 рН; 5,5 — 7,5 рН; 3 — 8,5 рН; 9.75 — 14 рН; 0 — 14 рН; 3 — 12 рН; 1 — 12,5 рН; 0 — 13 рН; 1 — 13 рН; 0 — 12 рН; 12 — 14 рН; материал чувствительного элемента(индикаторная часть): «платина»; «золото»; «серебро»; «ртуть»; разъем, тип соединения (подключение датчика): «беспроводное соединение Bluetooth»; «BNC»; «BNC + Pin»; «3.5 мм»; «DIN»; «многоштырьковое, multi pin»; «штыревое»; «штеккер»; «разъем байонетного типа»; «наконечник под винт М4»; «специальный»; «заклепка»; Таблица электродов, используемых с промышленными приборами 2
| Тип прибора | Электродная система (датчик), используемая с прибором | Примечание | ||||
| Измеряемая величина | Ионоселективный электрод, измерительный | Редокс-метрический электрод | Электрод сравнения | |||
| АЖА-101М АЖА101.1М АЖА-101.2М | % О 2 , мг/л | |||||
| ДК-02М | ||||||
| t, º С | См. графу 6 | - | - | Датчик t встроен в измерительное устройство | ||
| И-160 И-160МП И-160.1МП | pH | ЭСКЛ-08М | - | - | ||
| ЭСЛ-43-07СР | - | ЭВЛ-1М3.1 | ЭС-71-11 (стерилизуемый) | |||
| ЭСЛ-63-07СР | - | ЭСП-31-06 (до 150 0 С) | ||||
| pX + , pX - pX ++ , pX -- | См. графу 6 | - | ЭВЛ-1М3.1 | ЭСЛ-51-07СР(pNa, pAg ), ЭА-2 ( pS), ЭМ-Cl-01, ЭА-2 ( pS), ЭМ-Cl-01, ЭМ-I-01 (ЭМ-CN-01), ЭМ-NO 3 -07СР, ЭС-10-07 (pNa), ЭСС-01(pAg) , электроды других типов, находящиеся в обращении | ||
| Концентрация ионов сХ | Тоже | - | ЭВЛ-1М3.1 | Значение сХ соответствует измеренному значению рХ, пересчитанному в един. сХ | ||
| Eh, мВ | - | См. графу 6 | ЭВЛ-1М3.1 | ЭТП-02,ЭЗ-01,ЭО-01,ЭВП-1 электроды других типов, находящиеся в обращении | ||
| t, º С | ТКА-7.1 | - | - | И-160 | ||
| ТКА-1000.1 | - | - | И-160МП, И-160.1МП | |||
| рН-150М рН-150МП | рН | ЭСКЛ-08М.1 | - | - | рН-150М | |
| ЭСКЛ-08М.1 | - | ЭВЛ-1М3.1 | рН-150МП | |||
| ЭСЛ-43-07СР | ||||||
| ЭСЛ-63-07СР | ||||||
| Еh, мВ | - | См. графу 6 | ЭВЛ-1М3.1 | Тоже, что для И-160 | ||
| t, º С | ТКА-8М | - | - | рН-150М | ||
| ТКА-7.2 | рН-150МП | |||||
|
рН-150МП держателем с ножевым устройством |
рН |
ЭСКЛ-08М.1 |
- |
- |
- |
|
|
t, º С |
ТКА-ЛМ-11 |
- |
- |
- |
||
|
рХ-150МП |
рН |
ЭСКЛ-08М.1 |
- |
- |
||
|
ЭСЛ-43-07СР ЭСЛ-63-07СР |
- |
ЭВЛ-1М3.1 |
ЭС-71-11, ЭСП-31-06 |
|||
|
pX + , pX - |
См. графу 6 |
- |
ЭВЛ-1М3.1 |
Тоже, что для И-160 |
||
|
pX ++ , pX -- |
||||||
|
Еh, мВ |
См. графу 6 |
- |
ЭВЛ-1М3.1 |
Тоже, что для И-160 |
||
|
t, º С |
ТКА-7,2 |
- |
- |
- |
||
|
Нитратомер pNO 3 -07 |
pNO 3 |
ЭМ-NO 3 -07СР |
- |
ЭВЛ-1М3.1 |
Допускаются нитратные электроды других типов (аналоги) |
|
|
Концентрация ионов cNO 3 |
Тоже |
- |
Тоже |
Значение cNO 3 соответствует измеренному значению pNO 3 , пересчитанному в единицах сХ |
||
|
t, º С |
ТКА-1000.1 |
- |
- |
|||
|
БАТ-15.2 |
рН;pX; Еh, мВ |
См. графу 6 |
См. графу 6 |
См. графу 6 |
Электродная система иономера, используемого для титрования |
|
|
БАТ-15.2МП |
||||||
|
ПУ-1 |
Концентрация |
- |
ЭВЛ-1М4 |
|||
Примечание.1. Вместо электрода ЭВЛ-1М3.1, при необходимости, может быть применен выносной проточный электрод сравнения, включающий электрод ЭХСВ-1. 2. Электрод ЭСС-01 обеспечивает также измерение концентрации сульфидов-ионов (S2-) в пределах от 0,32 до 32 г/л. 3. Для подсоединения выводов электродов, указанных в графах 6, к соответствующим входам конкретных приборов, при необходимости могут использоваться адаптеры, штеккеры (переходники), поставляемые по заявке заказчика
Читать больше..Электроды используемые с промышленными лабораторными приборами
- 20 января 2019
- рН рН измерение ионы водорода pH-электрод титрование pH электрод индикаторные электроды синтез вода аналитическая химия потенциометрия измерение ph колориметрия промышленные приборы
Категории электродов: «Ион-селективные электроды»; «pH-электроды»; «датчики ОВП редокс-электроды»; «датчики проводимости»; «электроды вспомогательные (сравнения)»; «датчики температуры»; «кислородные датчики» по назначению: «электроды лабораторные»; «электроды промышленные»; «электроды промышленно-лабораторные»; «электроды ионоселективные»; «электроды вспомогательные»; по областям применения: «пищевые продукты и сырьё»; «полевые измерения»; «сильные кислоты и основания»; «измерения в малых объёмах»; «общелабораторные»; «промышленные»; Фильтр характеристик: «HANNA электроды»; «HANNA edge»; «с микрочипом»; «с температурным датчиком»; «с Bluetooth беспроводные электроды»; рабочий диапазон рН: 2 — 10 рН; 4 — 7 рН; 7 — 9 рН; 4,5 — 9,5; 0 — 11 рН; 5 — 7,5 рН; 1 — 10 рН; 5,5 — 8,5 рН; 0 — 8,5 рН; 2 — 8,5 рН; 1 — 9 рН; 5,5 — 7,5 рН; 3 — 8,5 рН; 9.75 — 14 рН; 0 — 14 рН; 3 — 12 рН; 1 — 12,5 рН; 0 — 13 рН; 1 — 13 рН; 0 — 12 рН; 12 — 14 рН; материал чувствительного элемента(индикаторная часть): «платина»; «золото»; «серебро»; «ртуть»; разъем, тип соединения (подключение датчика): «беспроводное соединение Bluetooth»; «BNC»; «BNC + Pin»; «3.5 мм»; «DIN»; «многоштырьковое, multi pin»; «штыревое»; «штеккер»; «разъем байонетного типа»; «наконечник под винт М4»; «специальный»; «заклепка»;
Таблица электродов, используемых с промышленными приборами 1
|
Тип прибора |
Электроды, используемые с приборами / Electrodesusedwithdevices | |||
| ионоселективный | ед.изм. | редокс-метрический | электрод сравнения | |
| АН-7529М АН-7560М | 5М2.840.019 | рН | 5М2.840.072 | |
| АС-7932М | 5М2.840.074 | рН | 5М2.840.072 | |
| П-210 П-215И П-215М | Монтируются в арматуре ДПг-4М, ДМ-5М, используемой с преобразователем | ЭХСВ-1 (в системе выносного проточного электрода)* ЭВП-08 (погружнойнепроточный) / | ||
| ЭА-2 ЭМ-I-01 ЭМ-CN-01 ЭМ-Cl-01 ЭМ-NO 3 -07 ЭС-10-07 ЭСП-01-14 ЭСП-04-14 ЭСП-31-06 ЭСС-01 | pS pJ pCN pCl pNO 3 pNa рН рН рН pAg | ЭО-01 ЭПВ-1 ЭТП-02 | ||
| pNa-205 | ЭС-10-07 ЭСЛ-63-07СР ЭСЛ-43-07СР | pNa рН рН | ЭХСВ-1 ЭВП-08 | |
| СХ-2 | ЭЗ-01 ЭО-01 | ЭХСВ-1 ЭВП-8 ЭСП-04-14 (10) | ||
| СЦ-2 | ЭМ-CN-01 | рCN | ЭЗ-01 ЭА-2 | ЭХСВ-1 ЭВП-08 ЭСП-04-14 (4) |
* - при избыточном давлении рабочей среды от 0,2 до 0,6 МПа должен использоваться с регулятором давления (например, РДС-1)
Таблица электродов, используемых с промышленными приборами 2
| Тип прибора | Электродная система (датчик), используемая с прибором | Примечание | ||||
| Измеряемая величина | Ионоселективный электрод, измерительный | Редокс-метрический электрод | Электрод сравнения | |||
| АЖА-101М АЖА101.1М АЖА-101.2М | % О 2 , мг/л | |||||
| ДК-02М | ||||||
| t, º С | См. графу 6 | - | - | Датчик t встроен в измерительное устройство | ||
| И-160 И-160МП И-160.1МП | pH | ЭСКЛ-08М | - | - | ||
| ЭСЛ-43-07СР | - | ЭВЛ-1М3.1 | ЭС-71-11 (стерилизуемый) | |||
| ЭСЛ-63-07СР | - | ЭСП-31-06 (до 150 0 С) | ||||
| pX + , pX - pX ++ , pX -- | См. графу 6 | - | ЭВЛ-1М3.1 | ЭСЛ-51-07СР(pNa, pAg ), ЭА-2 ( pS), ЭМ-Cl-01, ЭА-2 ( pS), ЭМ-Cl-01, ЭМ-I-01 (ЭМ-CN-01), ЭМ-NO 3 -07СР, ЭС-10-07 (pNa), ЭСС-01(pAg) , электроды других типов, находящиеся в обращении | ||
| Концентрация ионов сХ | Тоже | - | ЭВЛ-1М3.1 | Значение сХ соответствует измеренному значению рХ, пересчитанному в един. сХ | ||
| Eh, мВ | - | См. графу 6 | ЭВЛ-1М3.1 | ЭТП-02,ЭЗ-01,ЭО-01,ЭВП-1 электроды других типов, находящиеся в обращении | ||
| t, º С | ТКА-7.1 | - | - | И-160 | ||
| ТКА-1000.1 | - | - | И-160МП, И-160.1МП | |||
| рН-150М рН-150МП | рН | ЭСКЛ-08М.1 | - | - | рН-150М | |
| ЭСКЛ-08М.1 | - | ЭВЛ-1М3.1 | рН-150МП | |||
| ЭСЛ-43-07СР | ||||||
| ЭСЛ-63-07СР | ||||||
| Еh, мВ | - | См. графу 6 | ЭВЛ-1М3.1 | Тоже, что для И-160 | ||
| t, º С | ТКА-8М | - | - | рН-150М | ||
| ТКА-7.2 | рН-150МП | |||||
|
рН-150МП держателем с ножевым устройством |
рН |
ЭСКЛ-08М.1 |
- |
- |
- |
|
|
t, º С |
ТКА-ЛМ-11 |
- |
- |
- |
||
|
рХ-150МП |
рН |
ЭСКЛ-08М.1 |
- |
- |
||
|
ЭСЛ-43-07СР ЭСЛ-63-07СР |
- |
ЭВЛ-1М3.1 |
ЭС-71-11, ЭСП-31-06 |
|||
|
pX + , pX - |
См. графу 6 |
- |
ЭВЛ-1М3.1 |
Тоже, что для И-160 |
||
|
pX ++ , pX -- |
||||||
|
Еh, мВ |
См. графу 6 |
- |
ЭВЛ-1М3.1 |
Тоже, что для И-160 |
||
|
t, º С |
ТКА-7,2 |
- |
- |
- |
||
|
Нитратомер pNO 3 -07 |
pNO 3 |
ЭМ-NO 3 -07СР |
- |
ЭВЛ-1М3.1 |
Допускаются нитратные электроды других типов (аналоги) |
|
|
Концентрация ионов cNO 3 |
Тоже |
- |
Тоже |
Значение cNO 3 соответствует измеренному значению pNO 3 , пересчитанному в единицах сХ |
||
|
t, º С |
ТКА-1000.1 |
- |
- |
|||
|
БАТ-15.2 |
рН;pX; Еh, мВ |
См. графу 6 |
См. графу 6 |
См. графу 6 |
Электродная система иономера, используемого для титрования |
|
|
БАТ-15.2МП |
||||||
|
ПУ-1 |
Концентрация |
- |
ЭВЛ-1М4 |
|||
Примечание.1. Вместо электрода ЭВЛ-1М3.1, при необходимости, может быть применен выносной проточный электрод сравнения, включающий электрод ЭХСВ-1. 2. Электрод ЭСС-01 обеспечивает также измерение концентрации сульфидов-ионов (S2-) в пределах от 0,32 до 32 г/л. 3. Для подсоединения выводов электродов, указанных в графах 6, к соответствующим входам конкретных приборов, при необходимости могут использоваться адаптеры, штеккеры (переходники), поставляемые по заявке заказчика
Читать больше..Потенциометрия, количественный анализ
- 17 января 2019
- потенциометрия электрод индикаторные электроды титрование рh раствор ионометрия электрод сравнения pH-электрод pH электрод pH-метра водородный показатель ионы водорода измерение ph
Потенциометрия, количественный анализ
Читать больше..Потенциометрия, группа методов количественного анализа
- 17 января 2019
- потенциометрия электрод индикаторные электроды титрование рh раствор ионометрия электрод сравнения pH-электрод pH электрод pH-метра водородный показатель ионы водорода измерение ph
Потенциометрия, группа методов количественного анализа
Потенциометрия, количественный анализ
метод определения различных физико-химических величин
электроаналитический метод количественного анализа
Процедура, потенциометрия
В настоящее время легко доступны потенциометрические индикаторные электроды, такие как стеклянные электроды для измерения pH и окислительно-восстановительные электроды. С помощью этих электродов pH или окислительно-восстановительный потенциал могут быть считаны немедленно. В дополнение к этим двум электродам существуют и такие, которые реагируют на активность ионных частиц, ионоселективные электроды.
При использовании электрода серебро / сульфид серебра ион галогенида , сульфида , цианида , тиоцианата или хромата может быть определен путем титрования отрегулированным раствором нитрата серебра. С помощью жидких мембранных электродов можно определять концентрации ионов калия , магния , кальция или нитрата . Также для комплексометрического титрования могут быть использованы ионоселективные электроды.
Титрантом в комплексометрических определениях служит главным образом этилендиаминтетрауксусная кислота (ЭДТА) .
Потенциометрия используется в двух методах:
- Метод индикации представляет собой особую форму титрования, для которой во время титрования регистрируется изменение электродвижущей силы, а затем определяется точка эквивалентности . Данная процедура используется всякий раз, когда обычное титрование не может быть выполнено. Это может быть в том случае, если нет подходящего индикатора или решение настолько облачно или окрашено, что точка перехода не может быть обнаружена.
- Метод определения основан на определении концентрации путем измерения связанного электрохимического потенциала; оценка производится на основе калибровочной прямой, созданной с той же измерительной установкой.
Экспериментальная установка, потенциометрия
Ранее гальванический элемент состоял из двух электродов и раствора образца в качестве электролита. Разница потенциалов двух электродов определяется как электродвижущая сила ЭМК: {\ displaystyle EMK = | E_ {1} -E_ {2} |} {\ displaystyle EMK = | E_ {1} -E_ {2} |} Для конструкции используется электрод, электрод сравнения, потенциал которого очень хорошо известен и постоянен. Обычно для этого используется электрод второго типа .
На практике это обычно каломельный электрод или серебряно-серебряный хлоридный электрод. Разность напряжений между электродом сравнения и измерительным электродом, погруженным в раствор, можно определить с помощью вольтметра.
От типа титрования зависит выбор измерительного электрода
- Окислительно-восстановительное титрование платинового электрода,
- комплексометрическое определение либо ионно-чувствительного электрода, либо модифицированного серебряного электрода,
- Кислотно-основное титрование стеклянного электрода ,
- Аргентометрия серебряный электрод.
Измерительный электрод погружается в раствор образца и должен реагировать на определяемый ион, то есть потенциал этого электрода должен зависеть от концентрации определяемого иона. Эта зависимость описывается уравнением Нернста . В ходе эксперимента необходимо следить за тем, чтобы измерение проводилось без тока, в противном случае потенциалы были бы повреждены электролизом . Поэтому для измерения использовалась схема компенсации напряжения .
Оценка проводится путем определения точки эквивалентности. Лучший способ определить точку эквивалентности - построить на диаграмме первую производную кривой титрования. Максимум указывает точку эквивалентности. Объем титруемого раствора в мл нанесен на ось х, а измеренный потенциал в мВ на оси у.
точка эквивалентности в титриметрическом анализе момент титрования, когда число эквивалентов добавляемого титранта эквивалентно или равно числу эквивалентов определяемого вещества в образце. В некоторых случаях наблюдают несколько точек эквивалентности, следующих одна за другой, например, при титровании многоосновных кислот или же при титровании раствора, в котором присутствует несколько определяемых ионов.
На графике кривой титрования присутствует одна или несколько точек перегиба, соответствующих точкам эквивалентности. Точкой окончания титрования (подобна точке эквивалентности, но не то же самое) считают момент, при котором индикатор изменяет свой цвет при колориметрическом титровании.
Аналитическая химия, потенциометрия
Качественный химический анализ катионов и анионов. Аналитические группы анионов
Как известно все окружающие нас предметы разнообразные тела, различные соединения, горные ископаемые состоят из различных химических элементов. Если у исследуемого продукта известна химическая формула, то обычно определяют примеси, например, Na 2 CO 3 обычно содержит примеси NaHCO 3, CaCO 3, NaCl. Если исследуемый продукт представляет собой неизвестное вещество (какой-то металл, неметалл, окисел, соль, кислота и т. п.) то устанавливают его химическую формулу.
Из химии 8-го класса вам известно, что вещества - это то, из чего состоят физические тела. Самые разные тела могут состоять из одного и того же вещества (пластмасса, стекло, металл).
Свойства веществ
Признаки, по которым различные вещества подобны или отличаются друг от друга называются свойствами веществ. Каждое вещество имеет свои физические и химические свойства. Под химическими свойствами понимают способность вещества при определенных условиях превращаться в другие вещества.
- Вода может превращаться в новые вещества: водород и кислород; в более сложных процессах фотосинтеза углекислый газ и вода превращаются в глюкозу и кислород. Вспомним понятия чистые вещества и смеси веществ.
- Первые имеют постоянные физические и химические свойства, например, некоторые металлы. Нужно различать употребление в быту термина чистый (чистый шоколад, бензин, вода и т. д.).
- Бытовое употребление термина «чистый» отличается от химического понятия «чистое вещество».
Примеси компоненты, однородные и неоднородные примеси
В природе вещества почти никогда не встречаются в чистом виде, а обычно в виде смесей, составные части которых называются компонентами и могут быть однородными и неоднородными (вода, воздух, растворы, бензин и кровь, бетон, краски, чернила, асфальт, нефть, молоко и т. п.).
В химии чистые вещества и степень чистоты веществ четко обозначаются в виде надписей на этикетках химических реактивов - химически чистый (х.ч.), ч. Д. а., о. ч., и т. д.
Эти буквы являются руководством для химика-аналитика при проведении анализа и указываются в методических руководствах и нормативных документах.
- Определение химического состава любого исследуемого вещества сводится к разделению смесей, выделению и определению составляющих его компонентов.
Для каждого вещества необходимо разрабатывать методы разделения, т. к. универсальных методов выделения компонентов не существует, хотя бы потому, что каждая составляющая, т. е. каждый компонент имеет свои химические свойства. Однако, методы разделения смесей определенного типа были разработаны очень давно (алхимики) и применяются сейчас, другие методы, например, неразрушающего контроля и физико-химические разработаны в настоящее время.
Для разделения неоднородных смесей используются фильтрование и отстаивание, для однородных - выпаривание, дистилляция или перегонка (получение дистиллированной воды). Метод исследования основанный на разложении данного сложного вещества на более простые составные части называется анализом. Например, химический анализ воды можно установить путем разложения электрическим током (через воду, в которую добавлен электролит NaCl) пропускают постоянный электрический ток, при этом выделяются газообразный водород и кислород, что является доказательством того, что вода состоит из водорода и кислорода
2Н 2 О = 2Н 2 +О 2 .
Синтез получение сложного вещества из более простых веществ
Противоположностью анализа в химии является синтез - получение сложного вещества из более простых, например, синтез аммиака из водорода и азота при определенных условиях (температуре и давлении) N2 . +3Н 2 ↔2 NH 3 . В аналитической химии пользуются методом синтеза для определения состава неизвестных соединений путем сравнения их свойств со свойствами аналогичных соединений, но полученных синтетическим путем.
- Синтезом воды из водорода и кислорода было доказано, что вода сложное соединение, однако, необходимо было сделать и анализ воды, чтобы окончательно убедиться, что вода состоит из этих 2-х элементов.
Таким путем синтеза и анализа был определен состав воды и можно сказать словами Ф. Энгельса, что «без анализа нет синтеза» (Анти-Дюринг). Т. о. аналитической химией называют науку о методах определения качественного и количественного состава веществ или их смесей по интенсивности аналитического сигнала.
Теоретические основы методов исследования химического состава веществ
- Аналитическая химия разрабатывает теоретические основы методов исследования химического состава веществ и их практического применения.
Обнаружение компонентов (или ионов), содержащихся в данном веществе
- Задача качественного анализа - обнаружение компонентов (или ионов), содержащихся в данном веществе. Количественный анализ - это определение содержания составных частей сложного материала, результаты которого выражают в массовых долях (%), например, химически чистый сульфат меди CuSO 4 5H 2 O содержит массовую долю меди 25,44%, а фосфоритная мука - промышленное удобрение - массовую долю Р 2 О 5 от 20 до 30%.
Основные методы аналитической химии
Исследования вещества всегда начинаются с его качественного анализа, т. е. из определения того из каких компонентов (или ионов) состоит это вещество.
Если состав вещества известен, то сразу приступают к количественному анализу, предварительно выбрав наиболее подходящий метод.
Теоретические основы химического анализа составляют следующие законы и теоретические положения:
- периодический закон Д.И. Менделеева;
- закон действующих масс;
- теория электролитической диссоциации;
- химическое равновесие в гетерогенных системах;
- комплексообразование;
- амфотерность гидроксидов;
- автопротолиз (водородный и гидроксидный показатели);
- ОВР.
Характеристику основных методов аналитической химии мы будем давать в процессе их изучения в разделе количественного анализа.
Методы аналитической химии химические, физические и физико-химические
В аналитической химии используются различные методы: химические, физические и физико-химические. Коротко дадим характеристику каждому типу методов.
Химические методы основаны на превращениях, протекающих в растворах с образованием осадков, окрашенных соединений или газообразных веществ. Химические процессы, используемые в целях анализа, называют аналитическими реакциями. Аналитическими являются реакции, которые сопровождаются каким-нибудь внешним эффектом, позволяющим установить, что химический процесс связан с выпадение или растворением осадка, изменением окраски анализируемого раствора, выделением газообразных веществ. Требования к аналитическим реакциям и их особенности можно свести к следующим положениям:
выполнение анализа «сухим» или «мокрым» способом (сухой способ - это пирохимические методы, от греч. «пир» - огонь), сюда следует отнести пробы на окрашивание пламени при сгорании исследуемого вещества на петле платиновой (или нихромовой) проволочки с получением в результате окрашенного в характерный цвет пламени; получение цветных стекол или перлов за счет сплавления исследуемого вещества в петле платиновой проволки с бурой Nа 2 В 4 О 7 10Н 2 О, хром дает зеленый, кобальт - синий, марганец - фиолетовый перлы, рассмотрение металлических «корольков», получающихся при прокаливании анализируемых минералов на древесном угле с помощью паяльной трубки; метод растирания твердого анализируемого вещества с твердым реактивом, например, при растирании смеси соли аммония с Са(ОН) 2 выделяется аммиак.
Анализ сухим способом применяется для экспресс-анализов или в полевых условиях для качественного и полуколичественного исследования минаралов и руд. Для проведения мокрого анализа исследуемое вещество должно быть переведено в раствор и в дальнейшем реакции идут как реакции обнаружения ионов;
Условия: температура, реакция среды и концентрация обнаруживаемого иона
аналитическая реакция должна протекать быстро и полно при соблюдении определенных условий: температуры, реакции среды и концентрации обнаруживаемого иона. При выборе реакции обнаружения ионов руководствуются законм действующих масс и представлениями о химическом равновесии в растворах. При этом выделяются следующие характеристики аналитических реакций: селективность или избирательность; специфичность; чувствительность. Последняя характеристика связана с концентрацией обнаоуживаемого иона в растворе и если реакция удается при низкой концентрации иона, то говорят о высокочувствительной реакции.
Если вещество малорастворимо в воде и осадок выпадает при его низкой концентрации, то это высокочувствительная реакция, если вещество хорошо растворимо и выпадает в осадок при высокой концентрации иона, то реакция считается малочувствительной. Понятие чувствительности относится ко всем аналитическим реакциям, каким бы внешним эффектом они не сопровождались. Порог чувствительности реакций характеризуют количественно при помощи обнаруживаемого минимума, т. е. наименьшего количества иона, которое удается обнаружить с помощью данной реакции.
Выражают этот показатель в миллионных долях грамма - микрограммах 10 -6 г.
Например, при обнаружении иона К + в виде гексахлорплатината (1V) К 2 [PtCl 6 ] обнаруживаемый минимум составляет 0,1мкг. В качественном анализе применяют только те реакции, обнаруживаемый минимум которых не превышает 50 мкг. Селективные или избирательные реакции дают сходный внешний эффект с некольким ионами.
Оксалат аммония образует осадки с катионами
Оксалат аммония образует осадки с катионами Са 2+ , Ва 2+ ,Sr 2+ , и др. Но чем меньше таких ионов, тем более выражена избирательность (селективность) реакции. Селективность реакции может быть повышена с помощью маскирующих средств, которые переводят мешающие ионы в малодиссоциирующие или комплексные соединения. Концентрация посторонних ионов в растворе сильно понижается и помехи устраняются. Специфической называют такую реакцию, которая позволяет обнаружить ион в присутствии других ионов. Однако таких реакций мало (иод и синее окрашивание при наличии крахмала, обнаружение ионов NН 4 реактивом Несслера, обнаружение железа тиоцианатом с получением кроваво-красного осадка роданида железа
[Fе(SСN)] 2+ ;
дробный и систематический анализ. Обнаружение ионов с помощью селективных и специфических реакций в отдельных порциях раствора, производимое в любой последовательности называют дробным анализом.
Такой тип анализа широко применяется в агрохимических и заводских лабораториях. Когда состав анализируемого вещества хорошо известен и требуется проверить отсутствие или присутствие тех или иных примесей. Если же используемые реакции не селективны, а мешающее действие посторонних ионов устранить не удается, то в этом случае применяют т. н. систематический ход анализа т. е. определенную последовательность выполнения аналитических реакций, при которой каждый ион обнаруживают после того, как будут обнаружены и удалены мешающие ионы. В таком анализе применяются не только реакции обнаружения отдельных ионов, но и реакции отделения их друг от друга, используя различия в их растворимости или летучести. Полноту удаления мешающего иона проверяют в каждом случае специальной пробой.
Например, необходимо определить присутствие в растворе иона Са 2+ , который хорошо осаждается ионами щавелевой кислоты, т. е. при взаимодействии с оксалатом аммония
СаС1 2 + (NН 4 )С 2 О 4 = СаС 2 О 4↓ + NН 4 С1.
Но, если в растворе присутствуют ионы Ва 2+ , то они также способны выпадать в осадок в виде оксалата бария, поэтому необходимо проверить в отдельной порции раствора их наличие реакцией с дихроматом калия, при этом выпадает желтый осадок хромата бария. Кальций же способен выпадать в осадок в виде хромата только при очень высокой его концентрации из растворов солей кальция, в обычных условиях хромавт кальция хорошо растворим в воде. Следовательно, при наличии ионов бария в растворе, чтобы удалить мешающие осаждению кальция ионы бария необходимо на весь раствор подействовать раствором дихроматом калия, последний раствор берется с избытком, полученный желтый осадок хромата бария отфильтровывают, еще добавляют раствор хромата или дихромата, для проверки полноты осаждения бария и затем открывают ионы кальция раствором оксалата.
Такие манипуляции в виде последовательных реакций называются систематическим ходом анализа.
Систематический и дробный анализ не противопоставляются друг другу, а дополняют друг друга и применяются для аналитической классификации ионов. В ходе систематического анализа ионы выделяют из сложной смеси не по одному, а небольшими группами, пользуясь их одинаковым отношением к некоторым реактивам. Реактивы, позволяющие выделить из сложной смеси группу ионов, называют групповыми.
Удобство применения групповых реагентов заключается в том, что они позволяют сложную аналитическую задачу разделить на несколько более простых.
Групповые реагенты
Применение групповых реагентов указывает на присутствие или отсутствие целой группы ионов. Раствор сульфида натрия являясь групповым реагентом может перевести в раствор осадки ртути, мышьяка, сурьмы и олова при одновременном присутствии сульфидов многих катионов. Существует несколько различных аналитических классификаций катионов.
При систематическом анализе последовательность выделения аналитических групп из раствора не соответствует их нумерации.
В кислотно-основной классификации первым удаляют ионы аммония, остальные катионы первой группы анализируют в последнюю очередь. Аналитическая классификация катионов тесно связана с электронными структурами ионов и некоторые отступления от закономерностей связаны с отнесением отдельных катионов к той или иной группе для удобства анализа.
Катион магния
Исходя из его положения в периодической таблице, следовало бы отнести к 2-ой аналитической групп
е Са 2+ , Ва 2+ , Sr 2+ ,
но магний осаждается в виде фосфата и карбонат магния растворяется в избытке солей аммония и поэтому при использовании гидрата окиси аммония для осаждения гидратов окислов металлов, ион магния остается в растворе, поэтому его относят к первой группе катионов в нескольких классификациях катионов. Аналитические методы подразделяют на макро-, микро-, полумикро- и ультрамикрометоды в зависимости от количества исследуемого вещества, объема и техники выполняемых операций. Макрометод (грамм-метод) осуществляется со сравнительно большими количествами веществ:
1-10г сухог материала или 10 -100 мл раствора для их обработки применяют фильтрацию и промывку на бумажных фильтрах.
В микрометоде (миллиграмм-метод) берут 1 мг сухого вещества или 0,1 мл раствора, т. е. в 100-1000 раз меньше предыдущего.
Микрокристаллоскопический метод, метод Т.Е. Ловица или капельный метод
Реакции выполняют микрокристаллоскопическим (метод Т.Е. Ловица) или капельным методом (на стеклянной пластинке или фильтровальной бумаге, метод Н.А. Тананаева). Первый метод заключается в рассматривании получаемых кристаллов под микроскопом, например, желтые октаэдры гексахлорплатината калия, сильно преломляющие свет. Полумикрометод (сантиграмм-метод) - берут около 50 мг сухого вещества или 1 мл раствора, этот метод требует специальной аппаратуры и навыков, он позволяет уменьшить расход реактивов, ускорить выполнение анализа, избежать загрязнения воздуха. Микроаналитические методы: ультрамикроанализ 10 -6 -10 -9 г или 10 -4 10 -6 мл, субмикроанализ (нанограмм-метод) -10 -9 -10 -12 или 10 -7 -10 -10 мл, субультрамикроанализ (пикограмм-метод) менее 10 -12 г или 10 -10 мл
Отдельные методы и приемы химического анализа были известны ещё в глубокой древности. Уже тогда могли проводить анализы лекарственных препаратов, металлических руд. Однако, как наука анал. химия начала складываться значительно позже - по мере развития производства. Английский ученый Роберт Бойль (1627 - 1691)считается основоположником качественного анализа.
Закон сохранения массы
Для аналитической химии важное значение имеет закон сохранения массы (1748), который гласит: «общая масса продуктов реакции равна общей массе веществ, вступивших в реакцию» - как частный случай закона сохранения материи - один из основных законов аналитической химии (в частности для расчетов в количественном анализе). Например, если из 100 г исходных продуктов получается 120 г новых продуктов, значит в реакции участвует ещё 20 г веществ из окружающей среды.
Закон сохранения массы - основа стехиометрии и многих количественных законов химии, научное объяснение которой привело к созданию атомно-молекулярной теории.
Стехиометрия от греч. sτοίcheion первоначально, основа, элемент … метрия, в химии учение о количественных соотношениях между массами (объемами) реагирующих веществ (простых и сложных). Стехиометрия включает вывод химических формул, составление уравнений химических реакций, расчеты, применяемые в химическом анализе.
Термин ввел И. Рихтер (1792 - 94), обобщивший результаты своих определений масс кислот и оснований при образовании солей. Правила стехиометрии лежат в основе всех расчётов, связанных с уравнениями химических реакций. Бергман Торнберн Улаф, Клаус Карл Карлович, Севергин В. М
ЗАКОН ДЕЙСТВУЮЩИХ МАСС КАК ТЕОРЕТИЧЕСКАЯ ОСНОВА КАЧЕСТВЕННОГО АНАЛИЗА ПРИ ИЗУЧЕНИИ ГЕТЕРОГЕННЫХ ПРОЦЕССОВ ГИДРОЛИЗА И АМФОТЕРНОСТИ
Зависимость скорости реакции от концентрации реагирующих веществ, сформулированная Гульдбергом Като Максимилианом (1836 - 1902) - норвежским физикохимиком и математиком и Петером Вааге (1833 - 1900) норвежским физикохимиком и минералогом в 1864 - 1867 г. г. получила название закона действующих масс: скорость химической реакции прямо пропорциональна произведению молярных концентраций реагирующих веществ, возведенных в степени их стехиометрических коэффициентов.
Для конкретных случаев гомогенных систем: NН 4 Сl + NаОН = NН 4 ОН + NаСl и 2КОН + Н 2 SО 4 = К 2 SО 4 + 2Н 2 О, т. е.
при взаимодействии этих веществ в растворах уравнения скоростей реакций пишут соответсвенно:
v = k [NН 4 С1] [NаОН] и v = k [КОН] 2 [Н 2 SО 4 ].
В гетерогенных системах с участием твердой фазы скоростьреакции не зависит от массы твердого вещества (при небольшой поверхности его), а изменяется лишь в зависимости от концентрации газообразных (или растворенных) веществ, например:
С + СО 2 = 2СО; v = k[СО 2 ].
Обратимые реакции подвижное и химическое равновесие
В случае обратимых реакций устанавливается подвижное и химическое равновесие, при котором в системе одновременно присутствуют как исходные, так и образующиеся вещества. Химическим равновесием называют такое состояние системы реагирующих веществ, при котором скорости прямой и обратной реакций равны.
Для системы mА + nВ = рС + qD скорость прямой реакции v1 = k1 [А] m [В] n , а скорость обратной реакции v2 = k2 [С] р [D] q .
При химическом равновесии v1 = v2 .
Поэтому можно написать k1 [A] m [B] n = k2 [C] р [D] q .
После преобразования получим k1 / k2 = ([С] р [D] q ) / ([A] m [В] n ). Но отношение 2-х постоянных величин k1 / k2 есть величина постоянная, которую обозначают через К и называют константой равновесия: К = ([С] р [D] q ) / ([A] m [В] n ).
Химическое равновесие молярные концентрации вещества
При химическом равновесии произведение молярных концентраций получающихся веществ (продуктов реакции), деленное на произведение молярных концентраций исходных веществ, представляет собой постоянную для данной реакции величину, называемую константой равновесия (значение концентрации каждого компонента возводят в степень, равную стехиометрическому коэффициенту его в уравнении реакции). Константа равновесия показывает, во сколько раз скорость прямой реакции больше скорости обратной реакции k1 / k2 при данной температуре и одинаковых концентрациях.
Если константа равновесия равна К = 1, то скорости прямой и обратной реакций приблизительно равны.
Если величина К> 1, то преобладает прямая реакция и динамическое равновесие сдвинуто вправо. При К< 1 идет преимущественно обратная реакция и равновесие смещено влево.
Для обратимой реакции N2 + 3Н 2 = 2NН 3 уравнение константы равновесия имеет вид К = [NН 3 ]2 / [N 2 ][Н 2 ]3 .
Добавление в систему одного из реагирующих веществ вызывает смещение (сдвиг) химического равновесия, т. е. изменение равновесных концентраций. Равновесие вновь установится, но уже при других новых концентрациях исходных веществ и получающихся продуктов. В этом случае обязательно повышается скорость той реакции, при которой прибавленное вещество расходуется. Чтобы достичь более полного смещения (сдвига) химического равновесия нужно действовать избытком реагента, вызывающего это смещение.
В 1884 был сформулирован общий закон смещения химического равновесия, согласно которому при внешнем воздействии на равновесную систему химическое равновесие смещается в сторону, противоположную этому воздействию (принцип подвижного динамического равновесия, принцип Ле Шателье Анри Луи, 1850 - 1936, французский физикохимик и металловед). Уравнение константы химического равновесия является математическим выражение закона действующих масс. Именно Ле Шателье, независимо от Ф. Габера нашел (1901) условия синтеза аммиака.
3/2Н 2 + 1/2N 2 = NН 3 , реакция идет с выделением тепла -46,2 кДж/моль.
При увеличении концентрации азота или водорода происходит сдвиг реакции в сторону уменьшения концентрации этих веществ, т. е. в сторону образования аммиака и, наоборот; повышение температуры сместит реакцию в сторону образования исходных веществ, т. к. реакция идет с выделением тепла, понижение температуры - в сторону продолжения реакции. Повышение давления способствует уменьшению числа молекул, т. е. увеличению продуктов реакции, т. к. слева 2-ве молекулы, справа одна; уменьшение давления смещает равновесие реакции в сторону увеличения исходных продуктов. Если в реакции участвует одинаковое число молекул, то увеличение давления не дает результатов: N2 + О 2 = 2NО.
При выводе из сферы реакции образующихся веществ реакция идет в сторону их образования: СН 3 СООН + СН 2 ОН = СН 3 СООСН 3 + Н 2 О; добавлением в среду серной кислоты добиваются протекания реакции до конца, т. к. серная кислота поглощает воду и выводит ее из реакции. Таким образом, Обратимыми реакциями можно управлять сдвигая константу химического равновесия в сторону образования необходимого продукта и превращая обратимую реакцию в необратимую, т. е. реакцию, которая протекает до конца в одном направлении и завершается полным превращением исходных реагирующих веществ в конечный продукт. Принципы необратимости реакций:
образующиеся продукты уходят из сферы реакции в виде осадка или газа: ВаС1 2 + Н 2 SО 4 = ВаSО 4 + 2НС1.
- . образуется малодиссоциированное соединение, например, вода.
- . реакция идет с большим выделением тепла, например, горение магния: Мg + ½ О 2 = МgО; - 602,5 кДж/моль.
Обратимые реакции не идут до конца и заканчиваются установлением химического равновесия, которое можно определить как такое состояние системы реагирующих веществ, при котором скорости прямой и обратной реакций равны между собой, т. е. прямая и обратная реакции не прекращаются и находятся в состоянии динамического равновесия. Видимых изменений реакции не происходит, концентрации веществ остаются постоянными и равновесными. Однако закон действующих масс и понятие о химическом равновесии применимы только к неэлектролитам и слабым электролитам в разбавленных водных (или неводных) растворах.
Слабые электролиты в концентрированных водных растворах и все сильные электролиты (кислоты, щелочи, соли) не вполне подчиняются закону действующих масс.
Закон действующих масс справедлив только для идеальных систем и идеальных растворов.
Идеальных растворов не существует.
Электролитическая диссоциация, свойство водных растворов проводить электрический ток
Цель работы - ознакомление с электрохимическим методом определения степени диссоциации электролитов, основанном на свойстве их водных растворов проводить электрический ток. Изучение зависимости степени диссоциации слабого электролита от концентрации раствора.
Электролитами называют вещества, диссоциирующие в воде других полярных жидкостях или расплавах на ионы. Растворы и расплавы электролитов проводят электрический ток. Распад молекул вещества на ионы называют электролитической диссоциацией. Перенос тока в растворах и расплавах электролитов осуществляется ионами, поэтому их в отличие от электронных проводников называют ионными проводниками или проводниками второго рода. К электролитам относят соли, кислоты, основания.
Для количественной характеристики электролитической диссоциации вводится понятие степени диссоциации а:
- а = с/с 0 ,
где с - концентрация продиссоциировавших молекул, моль/л; с о - исходная концентрация раствора, моль/л.
Все электролиты делят на сильные и слабые. К сильным относятся те, степень диссоциации которых равна единице, т.е. с = с о . Электролитическая диссоциация сильных электролитов протекает необратимо:
- HNO 3 → Н + + NO 3-
- NaCl→ Na + +С1 -
К сильным электролитам относятся практически все соли, гидроксиды щелочных и щелочно-земельных метгллов и некоторые кислоты (например, НС1, HNO 3 и H2 SO 4 ).
Степень диссоциации слабых электролитов меньше единицы (с < со). Их диссоциация протекает обратимо:
- СН3СООН ↔ СНзСОО − + Н + ;Н 2 СО 3 ↔ Н + + НСОз−
Константу равновесия электролитической диссоциации слабого электролита называют константой диссоциация. Например, при 298 К
- с -с +
-
К дСН3СООН =
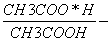 =1,8*10 -5
=1,8*10 -5 - c+ c-
-
К дН2СО3 =
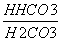 = 4,4.10 -7
= 4,4.10 -7
Сравнивая значения К д СН3СООН и К д Н2СО3" , можно сказать, что у угольной кислоты способность к диссоциации на ионы меньше, чем у уксусной.
Степень (а) и константа диссоциации (K д ) слабого электролита связаны зависимостью (закон Оствальда):
-
К д = а 2 со =_а 2 ____=
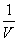 ,
, - (1-a) (1-a)V"
- где V = 1/с о - разведение раствора, л/моль.
В растворах электролитов осуществляются межионные взаимодействия, обусловленные силами притяжения и отталкивания. Наиболее заметны межионные взаимодействия в растворах сильных электролитов.
Для количественной характеристики межишных взаимодействий используется ионная сила раствора I (полусумма произведений концентраций всех ионов, присутствующих в растворе, на квадрат их заряда):
- I= 1/2∑с i zi2
Зная ионную силу раствора I, можно найти коэффициент γ, позволяющий определить некоторую величину а (активность), формально заменяющую концентрацию с:
- ai = γ i с i и γ, - коэффициент активности иона, который является функцией ионной силы раствора I и заряда иона zi .
Количественной характеристикой способности растворов электролитов проводить электрический ток является электрическая проводимость. Различают удельную 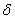 и молярную μ электрическую проводимость.
и молярную μ электрическую проводимость.
Удельная электрическая проводимость раствора (См • м -1 ) -величина, обратная удельному электрическому сопротивлению:
-
= 1/р (1)
Молярная электрическая проводимость раствора μ (См 2 *моль -1 ) - это электрическая проводимость такого объема раствора, в котором содержится один моль растворенного вещества:
-
μ =_ 1000
- с о (2)
μ возрастает с уменьшением с 0 (или с увеличением V), достигая некоторого предельного значения.
Проводниками электрического тока в растворах электролитов являются ионы, поэтому электрическая проводимость тем выше, чем больше ионов в растворе. Известно, что концентрация ионов в растворах слабых электролитов тем больше, чем больше степень диссоциации электролита а. Установлено, что между электрической проводимостью и степенью диссоциации существует взаимосвязь, вторая выражается формулой
-
а= μ/ μ
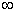
-
где μ - табличная величина (табулируются значения μ „ при 291 или 298 К).
Итак, чтобы вычислить степень диссоциации электролита (а) в растворе заданной концентрации при данной температуре, необходимо знать молярную электрическую проводимость (μ) этого раствора при той же температуре. Молярную электрическую проводимостъ рассчитывают по уравнению (2). Удельная электрическая проводимость раствора электролита, являясь величиной, обратной удельному сопротивлению раствора (р), может быть вычислена по экспериментально найденному значению электрического сопротивления (R) раствора электролита заданной концентрации при данной температуре.
Рис. 1. Ячейка для измерения электрического сопротивления раствора (а) и схема ее включения (б):
- пробки; 2 - кран для слива жидкости; 3 - платиновые электроды; 4-угольные электроды; 5 - исследуемый раствор; 6 - сосуд; 7 - источник | переменного тока; 8 - лампочка
Для измерения сопротивления раствора электролита при Т = const служит ячейка, изображенная на рис.1, а*. Через элекролит пропускают переменный электрический ток; схема включения ячейки в сеть приведена на рис. 1, б.
Сосуд 6 изготовляется из стеклянной трубки диаметром 12-15 мм и длиной 25-30 см. Концы трубки загнуты вверх. В горизонтальный участок трубки на расстоянии 10-12 см друг от друга впаивают два небольших кусочка платиновой проволоки (диам., 0,1 мм, длина 5-6 мм). К платиновым проволочкам припаивают тонкие медные проволочки, которые для большей прочности укрепляют на стеклянных выступах в нижней части сосуда. К ним припаивают провода, соединенные с вольтметром.
Платиновые электроды 3 служат для измерения падения напряжения в растворе электролита 5 в сосуде 6.
Ток подводится к раствору при помощи угольных электродов 4, закрепленных в резиновых пробках 1. Пробки вынимают при заполнении сосуда раствором; для сливания растворов служит кран 2. Через раствор пропускают переменный ток / (220 В). Для уменьшения тока в цепь последовательно с сосудом 6 включают дополнительное сопротивление - электрическую лампочку 8. Ток через раствор пропускают только во время измерений, чтобы не было разогревания раствора, изменяющего величину его электрического сопротивления.
Для измерения тока в цепи и падения напряжения между электродами 3 используют тестер (авометр), являющийся одновременно амперметром и вольтметром и позволяющий измерять как ток, так и напряжение в широком диапазоне их значений. Напряжение измеряется при параллельном включении вольтметра, а ток - при последовательном включении амперметра (рис. 1, б).
Измерив ток в цепи (7) и падение напряжения между электродами (U), можно вычислить сопротивление столба жидкости, заключенной между электродами:
- R = U/I, (4)
- и удельное сопротивление раствора электролита р:
- р = Rs/l, (5)
- где s - поперечное сечение проводника; l - его длина.
Отношение s/l называют "постоянной прибора". Эксперимент начинают с определения постоянной прибора. Для этого используют раствор электролита с известным удельным сопротивлением р (чаще всего 0,1 М раствор КС1, удельное сопротивление которого при 291 К и при 298 К равно соответственно 89,4 и 77,6 Ом*см). Измерив значения U и I в цепи, вычисляют R по уравнению (4), а затем постоянную прибора по уравнению (5).
Далее заменяют раствор хлорида калия исследуемым раствором и снова измеряют U и I. Зная постоянную прибора и сопротивление исследуемого раствора R, рассчитывают удельное сопротивление раствора электролита р и удельную электрическую проводимость а по уравнению (1).
Пользуясь соотношением (2), вычисляют μ исследуемого раствора.
Зная предельную молярную электрическую проводимость водного раствора исследуемого электролита μ , рассчитывают по уравнению (3) степень диссоциации а.
Солевой эффект, условия осаждения и растворения осадков.
Соли, кислоты, щелочи - электролиты, спирт этиловый, ацетон, диэтиловый эфир неэлектролиты. Теория электролитической диссоциации С. Аррениуса (1887):
.При растворении в воде электролиты частично или полностью распадаются на ионы и поэтому проводят электрический ток. Положительно заряженные ионы наз-ся катионами, отрицательно заряженные - анионами.
.Процесс диссоциации слабых электролитов - обратимый процесс, обратный процесс наз-ся ассоциацией или моляризацией
.В растворах электролитов ионы наход-ся в беспорядочном, хаотичном движении. При пропускании электрического тока их движение приобретает опред-ый порядок: катионы движутся к катоду, анионы - к аноду.
.Ионы по физическим, химическим и физиологическим свойствам отличаются от нейтральных атомов, из которых они образовались.
Пример ионы и атомы натрия и хлора. Ионы в растворе находятся в гидратированном состоянии, т.е. соединяются с молекулами воды (водород соединяется по донорно-акцепторному механизму,
образуя ионы гидроксония) [Na(H 2 O)] + , в обиходе мы пишем просто без молекул воды.
Сильные и слабые электролиты стр.18. Основания - это электролиты, которые при диссоциации образуют только один вид анионов - гидроксид-ионы - ОН - , кислоты образуют только один вид катионов - катионы Н + , в многоосновных кислотах диссоциация ступенчатая.-стр. 18. Амфотерные соединения диссоциируют по типу кислот и по типу оснований, т.к. прочность химических связей между атомами металла и кислорода и между атомами кислорода и водорода почти одинакова и в р-те в р-ре присутствуют и катионы Н + и анионы ОН - . Средние соли диссоциируют на катионы металла и анионы кислотного остатка, кислые соли диссоциируют на катион металла и сложный анион, в состав которого входит водород и кислотный остаток.
Гидрокарбонат - ион, который затем частично диссоциирует и дает анион СО 32- , двойная соль (образованная замещением водорода в многоосновной кислоте - сегнетова соль - тартрат калия магния
NаКС 4 Н 4 О 6 ,
алюмо-калиевые квасцы
КА1(SО 4 )12Н 2 О.
Факторы, влияющие на степень электролитической диссоциации
Природа электролита и растворителя: ионная связь в результате взаимодействия между молекулами растворителя и ионами электролита гидратирует. Это как бы первая стадия диссоциации, связи в кристаллической решетке ослабевают, кристаллическая решетка разрушается и в растворе находятся гидратированные ионы, окруженные гидратной оболочкой, состоящей из диполей воды - это хлористый натрий, хлористый калий, гидроксид бария.
Ионные соединения диссоциируют полностью. Соединения с полярной ковалентной связью диссоциируют частично, с присутствием в растворе ионов гидроксония - Н 3 О и зависит от диэлектрической постоянной растворителя, чем этот показатель больше, тем лучше происходит диссоциация. Например, серная к-та диссоциирует лучше в воде. Диэлектрическая постоянная которой равна 81, у этанола -27, у бензола -2.
- Концентрация раствора электролита: при повышении конц-ии, т.е. при частом столкновении ионов, усиливается образование молекул и степень диссоциации уменьшается, при разбавлении раствора - степень диссоциаци
.Повышение температуры усиливает степень диссоциации слабых электролитов, в связи с возрастанием движения ионов.
Электролиты в растворе
Влияние одноименного иона. Наличие в растворе 2-х электролитов, имеющих общий ион, наз-ый одноименным: ацетат натрия и уксусная к-та имеют одноименный анион ацетата. Слабые к-ты в присутствии своей соли или сильной к-ты почти не распадаются на ионы. Это видно из примера: добавление ацетата натрия к раствору уксусной к-ты увеличивает конц-ию одноименных ацетат-ионов. По принципу Ле-Шателье подобное действие сдвинет реакцию влево, т.е. в сторону образования уксусной к-ты. Добавление сильной к-ты сопровождается тем же эффектом за счет накопления катионов водорода, т.о. можно полностью подавить диссоциацию уксусной к-ты, стр.19. Степень диссоциации электролитов имеет большое значение в качественном анализе.
По величине степени диссоциации различных кислот можно предсказать растворение осадка в этих к-тах.
Так, оксалат бария не будет растворяться при комнатной температуре в уксусной к-те, т.к. эта к-та более слабая, чем щавелевая. Соляная и азотная к-ты вытесняют щавелевую к-ту из ее соли и растворяют оксалат бария. По этой же причине растворяются в соляной к-те ВаСrО4, СаС2О4, сульфид цинка, но не растворяются в уксусной к-те. С другой стороны для получения осадков стр. 19.
Силу электролита выражают другим, более удобным способом. Вместо константы диссоциации Кд, часто используют ее десятичный логарифм, взятый с обратным знаком:
рКд = -1g Кд.
Например, для уксусной к-ты Кд (СН3 СООН)=1,76∙10-5,
соответственно рКд (СН3СООН)=4,76,
где рКд наз-ся показателем константы диссоциации. Константа диссоциации и степень диссоциации - количественные характеристики процесса диссоциации. Между константой диссоциации и степенью диссоциации слабого электролита существует взаимосвязь. Для этого обозначим молярную концентрацию электролита, распадающегося на 2-ва иона, через с, а степень его диссоциации через α.
Тогда концентрация каждого из образующихся ионов будет равна сα, а концентрация недиссоциированных молекул с (1 - α). Подставив эти обозначения в уравнение константы диссоциации [А+][В-]/[АВ] = К, получим сα сα/с(1-α)=К, или они объединены законом разведения Оствальда:
Кд = α 2 с/(1- α), моль/л.
Это уравнение - математическое выражение закона разбавления Оствальда, который устанавливает зависимость между степенью диссоциации слабого электролита и его концентрацией. Величиной α в знаменателе пренебрегают, т.к. α ≤≤1, поэтому ур-ие принимает следующий вид: Кд = α 2с или α =√Кд /с Из данного ур-ия видно, что с увеличением концентрации α уменьшается, т.к. величина Кд при данной темп-ре остается постоянной величиной; при уменьшении концентрации электролита в р-ре степень его диссоциации увеличивается. рН и рОН буферных р-ов зависят от величины константы диссоциации кислоты или основания и от соотношения концентраций компонентов, что выражается уравнениями:
рН=рКа - lg С(кислота)/С(соль), или рОН=рКв - lg С (основание) /С(соль), где рКа и рКв - показатели константы диссоциации соответствующей кислоты и основания;
С(кислота) - концентрация кислоты; С(основание) - концентрация основания;
С(соль) - концентрация соли. Перевод одних малорастворимых электролитов в другие.
Согласно теории электролитической диссоциации все реакции в водных растворах электролитов являются реакциями между ионами и наз-ся ионными реакциями, а уравнения этих реакций - ионными уравнениями. При составлении ионных уравнений реакций следует руководствоваться тем, что вещества малодиссоциированные, малорастворимые (выпадающие в осадок) и газообразные записываются в молекулярной форме. Знак ↓, стоящий при формуле вещества, обозначает, что это вещество уходит из сферы реакции в виде осадка, знак ↑ означает, что в-во удаляется из сферы реакции в виде газа. Сильные электролиты как полностью диссоциированные записывают в виде ионов. Сумма электрических зарядов левой части уравнения должна быть равна сумме электрических зарядов правой части.
Пример: написать уравнения реакций между растворами хлорида железа (111) и гидроксида натрия в молекулярной и ионной формах. Разобьем решение задачи на 4-е этапа. 1-ый этап - уравнение в молекулярной форме: FeCl3 +3NaOH= Fe(OH)3↓+3NaCl. 2. Перепишем это уравнение, изобразив хорошо диссоциирующие вещества в виде ионов, а уходящие из сферы реакции - в виде молекул:
Fe3+ +3Cl- +3Na+ +3OH- = Fe(OH)3↓ +3Na+ +3Cl-.
Это ионное уравнение реакции. 3. Исключим из обоих частей ионного уравнения одинаковые ионы, т.е. ионы не участвующие в реакции (они подчеркнуты): Fe3+ +3Cl- +3Na+ +3OH=Fe(OH)3↓ +3Na+ +3Cl-. 4.
Уравнение в окончательном виде: Fe3+ +3OH- =Fe(OH)3↓.
Написать уравнения реакций между растворами хлорида калия и нитрата натрия. Уравнение записывается только в виде первых двух этапов, т. к. все соединения хорошо растворимы в воде и из сферы реакции не уходят, данная реакция обратима.
С точки зрения теории электролитической диссоциации реакция не происходит и уравнения следующих этапов написать нельзя.
Ионными уравнениями могут быть изображены любые реакции, протекающие в растворах между электролитами. Если в ходе таких реакций не происходит изменений зарядов ионов (не изменяется степень окисления), то они наз-ся ионообменными и сопровождаются (в случае их необратимости) образованием осадков - AgNO3 +HCl = AgCl↓ +HNO3; Ag+ + NO3- +H+ + Cl- =AgCl↓+ H+ + NO3-; Ag+ + Cl- =AgCl↓ Образованием газообразных малорастворимых веществ - Na2CO3 +H2SO4 =Na2SO4 +CO2↑+H2O; CO32- +2H =CO2↑ + H2O.
Образованием малодиссоциирующих веществ (слабых электролитов) HCl +KOH = KCl + H2O; H+ +OH- =H2O и образованием комплексных соединений (ионов) - CuSO45H2O+4NH3 =[Cu(NH3)4]SO4 + 5H2O; [Cu(H2O)4]2+ +4NH3= [Cu(NH3)4]2+ + 4H2O; [Cu(H2O)4]2++SO42-+ H2O+4NH3 = [Cu(NH3)4]2++ SO42-+5H2O.
Т.о., реакции обмена в растворах электролитов практически необратимо протекают в сторону образования осадков (малорастворимых веществ), газов (легколетучих веществ), слабых электролитов (малодиссоциирующих соединений) и комплексных ионов. Водородный показатель - рН; буква «р» - начальная буква датского слова potenz - математическая степень.
Выводы: 1. Электролиты в растворе или расплаве диссоциируют на ионы, благодаря чему и проводят электрический ток.
Чем больше ионов в растворе, тем выше его электрическая проводимость. Главные ион образующие вещества - кислоты, основания, соли.
.Ионы в водных растворах гидратированы, т.е. химически связаны с ионами воды. Гидратация ионов - основная причина диссоциации.
.С помощью теории электролитической диссоциации дается определение кислотам, основаниям и солям: кислоты выделяют в раствор ионы водорода, основания - гидроксид ионы, а соли - катионы металлов (и аммония) и анионы кислотных остатков
.Различают сильные и слабые электролиты. Сильные электролиты полностью диссоциируют на ионы, концентрация ионов в растворе велика. Слабые электролиты лишь частично диссоциируют на ионы и концентрация ионов в растворе не достигает значительных величин.
.Вода - слабо диссоциирующее соединение, в результате образуются ионы водорода и гидроксид-ионы. Т.к. количество этих ионов равнозначно, т.е. эквивалентно, реакция среды нейтральна и имеет рН 7. Для кислых растворов рН< 7, у щелочных рН>7. Схема зависимости между концентрацией ионов водорода, величиной рН и реакцией раствора, стр. 107 Химия, Г.П. Хомченко, для подготовительных отделений, ВШ.
.Количественные характеристики процесса диссоциации: степень электролитической диссоциации или ионизации, α - число, показывающее какая часть от общего количества электролита распадается на ионы; Кд - константа диссоциации, характеристика силы электролитов; f - коэффициент активности иона, i - ионная сила раствора, i =0,5∑ci zi2.
.В водных растворах электролитов реакции протекают между ионами и изображаются ионными уравнениями. Они протекают практически необратимо в сторону образования осадков (малорастворимых веществ), газов (легколетучих веществ), слабых электролитов (малодиссоциирующих соединений) и комплексных ионов. В аналитической химии используются реакции для осаждения осадков с помощью введения в раствор электролита, содержащего одноименные с осадком ионы и введение в раствор сильного электролита с посторонним ионами для растворения осадка - солевой эффект.
.Согласно протолитической теории кислотой называется любой донор протона, а основанием - акцептор протона. Т.е. кислоты - вещества, молекулы или ионы, отщепляющие при данной реакции протоны. Основания - вещества, молекулы или ионы, присоединяющие протоны, эти соединения получили общее название протолиты, в соответствии с протолитической теорией кислот и оснований датского ученого Йоханнеса Николауса Бренстеда и английского ученого Томаса Мартина Лоури (1928-1929). Эта теория объясняет процессы, протекающие в неводных растворах.
Соль хлорид аммония в водном р-ре, то в жидком аммиаке - это к-та, растворяет металлы с выделением водорода; азотная к-та в жидком фтороводороде или безводной серной к-те является основанием. Реакция отщепления протона: Кислота↔основание +Н+. Кислота и основание такого процесса наз-ся сопряженными. Это кислотно-основная пара. Свободные протоны в растворах самостоятельно не существуют, они переходят от к-ты к какому-либо основанию.
Поэтому в растворе всегда протекают 2-ва процесса: Кислота1↔ Основание1 +Н+ и Основание2+Н+ ↔Кислота2. Соответственно, равновесие между ними изображается уравнением: Кислота1 + Основание2 ↔ Основание1 +Кислота2. Например, реакцию нейтрализации уксусной к-ты аммиаком можно представить так: СН3СООН↔СН3СОО- +Н+; NH3 +H+↔NH4+; и общее уравнение CH3COOH (кислота1)+ NH3 (основание2)↔CH3COO- (основание1) + NH4+(кислота2).
Здесь уксусная к-та является кислотой (отдает протоны Н+), аммиак - основанием (принимает протоны), СН3СОО+- ионы играют роль основания (в этом обратимом процессе они могут принять протоны и превратиться в уксусную к-ту), а ионы аммония играют роль кислоты (могут отдать протоны). Аналогично, реакция проявления основных свойств азотной к-ты, растворенной в жидком HF: HF(кислота1)+HNO3 (основание2)↔F_ (основание1) +[H2NO3]+ (кислота2).
Основные положения «общей» или «расширенной», теории кислот и оснований:
- кислота является донором, а основание - акцептором протона;
- кислоты и основания существуют только как сопряженные пары;
- протон не существует в р-ре в свободном виде, в воде он образует ион ОН3+.
Протолитическая теория расширила круг оснований и кислот по сравнению с представлениями тории электролитической диссоциации: ими могут быть и ионы.
Одно и то же вещество в зависимости от того, с чем оно реагирует может быть или кислотой или основанием.
Теория вскрыла относительность понятий о кислотах и основаниях и показала, что продукты их взаимодействия следует рассматривать как новые кислоты и основания.
Протолитическая теория является современной и более общей теорией кислот и оснований.
- Уравнение электролитической диссоциации воды.
- Константа диссоциации воды - ионное произведение воды.
- Водородный показатель - рН.
- Амфотерность и ее значение в химическом анализе.
По уравнению электролитической диссоциации воды видно, что вода типичный электролит, проявляющий амфотерные свойства: Н2О <-> 2Н+ + ОН- или Н2О + Н2О <-> Н3О+ + ОН-. Однако, для простоты вместо иона гидроксония, в виде которого водород находится в растворе, пишут и говорят об ионе водорода (такая замена не влияет на расчет). Т. о. при диссоциации воды образуются ионы водорода, понимаемые как ионы гидроксония - носители кислотных свойств и ионы гидроксила - обуславливающие щелочные свойства воды. Как видно из уравнения диссоциации в жидкой воде концентрация ионов водорода приблизительно равна концентрации ионов гидроксила и поэтому можно написать, что в воде [Н+] = [ОН-].
Применив закон действующих масс к равновесной системе электролитической диссоциации воды получим: [Н+][ОН-] / [Н2О] = К, т. е. получаем константу электролитической диссоциации воды. Подобные процессы называют реакциями автопротолиза, в этих реакциях одна молекула амфипротного растворителя ведет себя как кислота, а другая как основание. Вычисления по электрической проводимости показали, что концентрация ионов водорода и гидроксид-ионов в воде оказалась равна 10-7 моль/л при 220 С. Преобразуя это уравнение, получаем [Н+][ОН-] = [Н2О] К. Но степень диссоциации воды очень мала, и концентрацию ее недиссоциированных молекул в любом разбавленном водном растворе можно считать величиной постоянной.
Т. о. в правой части уравнения находятся 2-ве постоянные величины: [Н2О] - концентрация недиссоциированных молекул воды и К - константа диссоциации. Но произведение 2-х постоянных величин есть также величина постоянная. Поэтому заменив [Н2О]К новой константой, получим [Н+] [ОН-] = КН2О. Следовательно, как бы не изменялись концентрации ионов Н+ и ОН- в воде или в разбавленном водном растворе, произведение их остается величиной практически постоянной. Эту величину называют ионным произведением воды.
Числовое значение этой константы нетрудно найти, подставив в уравнение значения концентраций водородных и гидроксид-ионов в воде:
КН2О = [Н+][ОН-] = 10-710-7 = 10-14.
Реакцию того или иного раствора принято характеризовать концентрацией водородных ионов, т.к. концентрацию ионов ОН- легко вычислить, исходя из ионного произведения воды. Концентрация ионов Н+ и ОН- изменяется в обратно пропорциональной зависимости, но никогда не становится равной нулю: Н+= КН2О / ОН- или ОН- = КН2О/Н+. Т. к. ионное произведение воды при данной температуре неизменно и равно 10-14, а величины [Н+] и [ОН-] переменны, то по этим их величинам можно судить о кислотности или щелочности раствора. Реакция среды кислая, если величина [ОН-] уменьшается, т. е. становится меньше 10-7 г-ион/л; реакция щелочная при соответственном увеличении концентрации ионов ОН-.
Следовательно, всякий раствор будет называться нейтральным, если в нем при комнатной температуре [Н+] = [ОН-] = 10-7 г-ион/л.
- Для кислых растворов справедливо условие [Н+]>[ОН-]; [Н+]>10-7 г-ион/л, [ОН-]<10-7.
- Для щелочных растворов справедливо - [Н+]<[ОН-]; [Н+]<10-7, [ОН]<10-7 г-ион/л.
Концентрация ионов водорода в растворе
Например, если концентрация ионов водорода в растворе равна 10-3 г-ион/л, реакция среды кислая, величину [ОН-] в таком растворе можно рассчитать из уравнения ионного произведения воды:
[Н+][ОН-] = 10-14г-ион/л, следовательно, [ОН-] =10-14/10-3=10-11 г-ион/л.
Ионизация воды эндотермический процесс, протекающий с поглощением тепла, согласно закону Ле-Шателье при повышении температуры равновесие ионизации смещается в сторону образования ионов, что приводит к увеличению ионного произведения воды. Зная экспериментальные табличные данные ионного произведения воды можно решать различные задачи. Однако, характеризовать основность и кислотность растворов концентрацией ионов водорода с отрицательным показателем степени оказалось практически неудобным.
И поэтому в практике применяют водородный показатель - рН, отрицательный логарифм концентрации ионов водорода: рН= -lg [Н+]. Т. о. нейтральная среда рН=7, кислая рН<7, щелочная рН>7. Чем меньше величина рН, тем больше концентрация водородных ионов, тем больше кислотность раствора. Кислотность раствора растет с уменьшением рН, в то время как щелочность возрастает с его увеличением. Все сказанное наглядно видно, если изобразить значения рН в виде схемы. Конечные точки этой схемы соответствуют, примерно, концентрации ионов водорода 1н. раствора НС1 (рН=0) и 1н. раствора NаОН (рН=14). Конечно могут быть более кислые растворы с рН<0 и более щелочные с рН >14, но в этих случаях кислотность или щелочность этих растворов принято выражать в молях кислоты или щелочи на литр раствора.
Наряду с водородным показателем нередко применяют гидроксильный показатель рОН= -lg [ОН-].
Если уравнение ионного произведения воды [Н+][ОН-] = 10-14 прологарифмировать, а затем поменять у логарифмов знаки на обратные, то получим рН + рОН= рКН2О=14, где рКН2О= --lgКН2О=-lg10-14=14.
Отсюда следует, что в любом водном растворе действительно соотношение рН= рКН2О - рОН = 14 - рОН (при 250 С).
Т. о., используя эти уравнения можно решать различные задачи: вычислять рН растворов кислот и оснований, вычислять концентрацию ионов Н+ и ОН- по известным значениям рН и обратно. При расчете водородного показателя среды водных растворов сильных кислот и оснований следует учитывать необратимость процесса их электролитической диссоциации (α=1). Концентрация ионов Н+ в растворах одноосновных сильных кислот и концентрация ионов ОН- в растворах одноосновных щелочей численно равна молярности растворов.
Концентрация ионов, например
Н+ в 0,001М растворе НС1 равна 0,001 моль/л, а концентрация ОН- в 0,01 М растворе NаОН равна 0,01 моль/л.
Для успешного проведения качественных реакций в нужном направлении необходимо соблюдение определенных условий, в первую очередь, определенной величины рН исследуемого раствора. Если требуемая величина рН не соблюдается, как правило, реакция не идет в нужном направлении и всю работу приходится начинать с начала.
Так, например, катион магния отнесен к 1-ой группе катионов (по кислотно-основной схеме определения катионов) и его можно осадить из раствора, содержащего ион магния в виде гидроокиси, согласно уравнению Мg2++2ОН--->Мg(ОН)2. Однако эта реакция идет только при определенном значении рОН-<14, т. е. при рН >10. При добавлении к раствору соли магния гидроокиси аммония осадок Мg(ОН)2 не выпадает даже при рН=9 или осаждение бывает неполным. Полное осаждение происходит при действии сильных оснований, а в присутствии солей аммония гидрат окиси магния Мg(ОН)2 остается в растворе. Поэтому магний относят к первой группе катионов.
Учитывая вышеизложенное, при проведении качественных реакций необходимо указывать не только характер среды: «кислая» либо «щелочная», но и точное значение рН. Методы определения рН среды: колориметрический и потенциометрический. Наиболее точный потенциометрический метод, который осуществляется с помощью прибора потенциометра или иономера.
Прибор необходимо предварительно подготовить и произвести настройку с помощью тест-стандартов или нормодоз, т. е. растворов с известным значением рН, измерение рН осуществляется с помощью электролитической ячейки и двух электродов. Колориметрический метод основан на применении реактивов, изменяющих свою окраску в зависимости от концентрации водородных ионов, которые называются индикаторами Это слабые органические кислоты или основания недиссоциированные молекулы которых имеют различную окраску, они называются кислотно-основными индикаторами. Лакмус, например, содержит азолитминовую кислоту недиссоциированные молекулы которой красного цвета, а анионы синего.
Добавление к раствору кислоты или щелочи изменяет рН раствора с чем и связано изменение окраски, рассматриваемых индикаторов. Важным условием соблюдение значений рН является при определении амфотерных катионов 4-ой группы. - lg [Н+]- -lg [ОН-]= lgКН2О = 14.
Для приблизительного определения рН среды в ходе качественного анализа чаще всего пользуются следующими индикаторами: метиловым оранжевым, метиловым красным, лакмусом, фенолфталеином, феноловым красным, малахитовым зеленым и др. К отдельным пробам испытуемого раствора добавляют по 1 - 2 капли раствора каждого индикатора. При этом нужно знать какую окраску имеет используемый индикатор в зависимости от рН среды.
Например лакмус (или лакмусовая бумажка) краснеет в кислой среде при рН <5, а синеет в щелочной среде при рН>8.
Для фенолфталеина малиновая окраска характерна в щелочной среде (при рН>10; начиная от рН 8,2 и меньше он бесцветен.
Универсальные индикаторы, например, ЗИВ-1, представляет собой сухую смесь из 5-и индикаторов: диметиламиназобензола, бромтимолового синего, метилового красного, фенолфталеина и тимолфталеина. Готовая смесь в ампулах, в количестве 0,16 г растворяется в 100 мл 80 %-го спирта. В зависимости от рН раствора универсальный индикатор приобретает окраску в соответствии со шкалой. Действие одноименных ионов. Степень электролитической диссоциации вещества зависит не только от его концентрации в растворе, но и от добавления в раствор других электролитов. Степень диссоциации слабого электролита можно понизить, добавляя в раствор сильный электролит, содержащий ион, одноименный с первым электролитом.
Это явление объясняется с точки зрения закона действующих масс и константы равновесия реакции диссоциации. Предлагается самим студентам объяснить изменение степени диссоциации раствора уксусной кислоты при добавлении уксуснокислого аммония или натрия и раствора аммиака при добавлении раствора хлорида аммония. Т. о. степень диссоциации слабого электролита понижается при введении в раствор какого-нибудь сильного электролита содержащего одноименный ион.
Растворы, рН которых почти не изменяется от прибавления небольших объемов сильных кислот или щелочей, а также от разбавления называют буферными растворами или буферными смесями. Они представляют собой смеси электролитов, содержащие одноименные ионы. Например, ацетатный буферный раствор - это смесь уксусной кислоты и ацетата натрия, аммонийный буферный раствор - смесь гидрата окиси аммония и хлористого аммония. Ацетатная смесь проявляет свое буферное действие только до тех пор, пока концентрация прибавленной кислоты или щелочи не превысит приблизительно 0,08 моль/л. Количество молей сильной кислоты или сильного основания, прибавление которого к 1 л буферного раствора изменяет его рН на единицу, характеризует буферную емкость раствора.
Буферная емкость измеряется количеством г-эквивалентов или моль/л сильной кислоты или сильного основания
Следовательно, буферная емкость измеряется количеством г-экв или моль/л сильной кислоты или сильного основания, которое необходимо добавить к 1 л буферной системы, чтобы изменить его значение рН на единицу. Буферным действием обладают также смеси кислых солей с различной замещенностью водорода металлом. Например, в буферной смеси дигидрофосфата и гидрофосфата натрия первая соль играет роль слабой кислота, а вторая - роль её соли.
Фосфатный буферный раствор (NаН2РО4 + NаНРО4) поддерживает рН 6,8. Т. о. варьируя концентрации слабой кислоты и её соли, можно получить буферные растворы с разными значениями рН.
Примерами буферных действий в природе является кровь, лимфа, почва. Для вычисления рН в буферных смесях применяют уравнение рН = рКк - lg(ск/сс), где рК является силовым показателем кислоты или это логарифм константы диссоциации кислоты; ск - концентрация кислоты, а сс - концентрация соли. Указанное уравнение получено после логарифмирования уравнения константы диссоциации слабой кислоты, входящей в буферную смесь.
Для смесей слабых оснований и их солей рОН = рКо - lg(со/сс). Учитывая, что рН = рОН = 14, можно написать рН = 14 - рОН = 14 - рКо + lg(со/сс). Т.о. 1. мах буферная емкость набл-ся у смесей содержащих равные концентрации компонентов; 2. По мере добавления к буферному раствору кислоты или щелочи устойчивость его к изменению рН постепенно уменьшается. 3. Буферная емкость раствора тем больше, чем выше концентрация компонентов буферной смеси. При проведении анализов при постоянном рН следует учитывать буферную емкость используемой смеси.
Лекция Сущность гидролиза и типы гидролиза солей. Как известно, растворы средних солей имеют различную реакцию среды: кислую, щелочную, нейтральную, хотя в их состав не входят ни гидроксильные, ни водородные ионы. Отчего это происходит? Объяснение этого явления следует искать в процессе взаимодействия солей с водой. Гидролиз - это разложение вещества водой или гидролизом соли наз-ся взаимодействие ионов соли с ионами воды, которое приводит к образованию слабых электролитов. Гидролиз соли - это реакция, обратная реакции нейтрализации. Поэтому каждую соль можно представить себе как соединение, образованное основанием и кислотой, которые бывают сильными и слабыми электролитами.
При этом различают 4-е типа солей. Соли, образованные слабой к-ой и слабым основанием при гидролизе сообщают раствору реакцию среды, зависящую от степени диссоциации продуктов гидролиза - кислоты и основания; если преобладают ОН- ионы, реакция среды щелочная, если ионы Н+ - кислая, если же их число одинаково, то среда нейтральная. Например: соль CH3COONH4. Ионы этой соли одновременно связывают ионы Н+ и ОН-, смещая равновесие диссоциации воды:
CH3COO-+ NH4+ +H2O→←CH3COOH+NH4OH (NH3H2O), среда нейтральная, стр. 23.
Однако, реакции водных растворов
(NH4)2S и (NH4)2CO3
имеют слабо щелочную -9,2 реакцию, т.к. степень диссоциации NH4OH, больше степени диссоциации ионов НСО3- и S2-. Уравнение гидролиза лучше писать в сокращенной ионной форме, вместо формулы гидроксида аммония лучше писать формулу гидрата аммиака. Обратимый и необратимый гидролиз.
Для большинства солей гидролиз процесс обратимый.
Когда продукты гидролиза уходят из сферы реакции, гидролиз протекает необратимо, например: Al2 S3 + 6H2O=2Al(OH)3↓ + 3H2S (в уравнения необратимого гидролиза ставится знак равенства). Гидролиз, в р-те которого образ-ся малорастворимые или газообразные продукты, удаляющиеся из сферы реакции, являются необратимыми. Степень гидролиза, смещение равновесия гидролиза. Влияние различных факторов на смещение равновесия гидролиза используется в аналитической химии для обнаружения отдельных ионов, регулирования кислотности и щелочности анализируемых р-ов, разделения ионов при систематическом качественном анализе.
Гидролиз солей происходит тогда, когда их ионы, образующиеся в процессе электролитической диссоциации, способны образовывать с водой слабые (малодиссоцированные) электролиты. Гидролизом называется реакция обменного разложения между водой и соответствующим соединением.
Механизм гидролиза солей, распадающихся в воде на ионы, определяется взаимодействием ионов с их гидратной оболочкой. Характер и степень распада гидратной оболочки зависят от поляризующего действия ионов: чем сильнее это действие, тем интенсивнее протекает гидролиз. Катионы связаны с молекулами воды донорно-акцепторной связью. Акцепторная способность катионов возрастает с увеличением их заряда и уменьшением размеров. Слабые акцепторы (катионы щелочных и щелочноземельных металлов) практически не поляризуют связь О - Н координированной молекулы воды.
Разложение воды в этом случае не происходит: Na+ + HOH ↔ реакция не идет. 2-х и 3-х зарядные катионы
(Cu2+, Fe2+, Fe3+, Al3+ и др.)
разлагают молекулы воды с образованием гидроксо-аквакомплексов. Образуется избыток ионов гидроксония (Н3О), т.е среда становиться кислой. Гидратация анионов осуществляется за счет водородной связи. Чем больше отрицательный заряд и меньше размер аниона, тем легче протон отрывается от молекулы воды. Оторвавшийся протон присоединяется к аниону. Слабые доноры электронных пар - однозарядные ионы (Cl-, Br-, I-, NO3- и др.), а также ионы SO42-, SiF62- не вызывают заметного разложения воды. Cl- +HOH ↔ реакция не идет.
Доноры средней силы -2-х и многозарядные анионы
CO32-, PO43- CN- -
вызывают обратимое разложение воды: CO32- + HOH↔HCO3- +OH- -появляется избыток ионов ОН-, т.е. среда становится щелочной. Т.о. суммарный эффект гидролиза солей определяется характером катионов и анионов.
- Гидролиз практически не идет, рН среды не изменяется, если соль при ионизации образует малополяризующиеся катионы и малополяризующиеся анионы: NaCl↔ Na+ + Cl-, Na+ + HOH↔реакция не идет.
. Гидролиз приводит к образованию кислой среды, если соль при ионизации образует среднеполяризующиеся катионы и малополяризующиеся анионы:
CuCl2↔Cu2++ 2Cl-, Cu2+ +HOH↔CuOH+ + H+, Cl- + HOH↔ реакция не идет, в молекулярной форме CuCl2+ HOH↔Cu(OH)Cl + HCl.
. Гидролиз приводит к образованию щелочной среды, если соль при ионизации образует слабополяризующиеся катионы и среднеполяризующиеся анионы:
Na2CO3↔2Na+ +CO32-, 2Na++ HOH↔реакция не идет, CO32- +HOH↔HCO3-+OH-, в молекулярной форме Na2CO3+HOH↔NaHCO3 + NaOH.
. Полный гидролиз с образованием малорастворимых оснований и слабых кислот происходит, если соль при диссоциации образует среднеполяризующиеся катионы и анионы: А1(СО3)3+6Н2О↔2А1(ОН)3+ 3Н2СО3.
Процесс гидролиза при необходимости можно подавлять введением реагента, имеющего одноименные ионы с продуктами гидролиза или изменяя температуру:
SbCl3 + 2H2O = Sb(OH)2Cl + 2HCl; Sb(OH)2Cl=SbOCl↓+H2O.
Хлорид сурьмы в воде гидролизуется с образованием малорастворимого оксохлорида сурьмы, выпадающего в осадок. Для подавления гидролиза в этом случае в раствор добавляют НС1, которая смещает равновесие гидролиза влево, что позволяет избежать выпадения осадка и сурьма в растворе присутствует в виде растворимых хлоридных комплексов. Иногда необходимо увеличить глубину гидролиза. Степенью гидролиза h называется величина, равная отношению прогидролизовавшихся ионов nh к общему числу n исходных ионов h=nh/n, (в долях единицы или в процентах), она увеличивается с уменьшением концентрации раствора и с ростом температуры. В растворах солей, при гидролитических процессах устанавливается гидролитическое равновесие и константа гидролитического равновесия Кh наз-ся концентрационной константой гидролиза и выражается в общем случае реакции
В- + Н2О=НВ +ОН- как отношение Кh =[HB][OH- ]/[B-].
При проведении соответствующих преобразований получают, что Кh=Kw/Ka, т.о. константа гидролиза равна константе основности основания, сопряженного с кислотой, анион которой гидролизуется. Обычно гидролизуется лишь очень малая часть ионов, образующихся при электролитической диссоциации соли, поэтому степень гидролиза намного меньше единицы: h<<1. Константа гидролиза соли слабой одноосновной к-ты и слабого основания равна Кгидр.= КН2О / Ккисл., где КН2О или Кw- ионное произведение воды [Н3О+][ОН-], а Ккисл. константа диссоциации слабой одноосновной кислоты. В результате всех преобразований можно рассчитать значение рН раствора, в котором гидролизуется анион слабой одноосновной к-ты по формуле рН=7 +0,5(рКа- рс b). рН гидролиза катиона слабого основания, например, а растворах солей аммония, содержащих катион аммония NH4+: NH4+ +H2O = H3O+ +NH3, раньше записывали NH4+ +H2O=NH4OH +H+.
В общем случае ВН+ +Н2О = В + Н3О+. И рН =7-0,5(рКb -рса). В общем случае уравнение гидролиза катиона В+ слабого основания ВОН: В+ +2Н2О=ВОН +Н3О+. При гидролизе соли ВА, образованной катионом В+ слабого однокислотного основания ВОН и анионом А- слабой одноосновной кислоты НА. Как сильный электролит соль ВА в водном растворе полностью распадается на ионы ВА→ В+ +А-, которые гидролизуются: В+ + 2Н2О = ВОН +Н3О+ и А- +Н2О= НА +ОН- расчет рН в этом случае по формуле: рН≈0,5(рКw +pKa - pKb), где Ka и Kb -соответственно константы ионизации слабой кислоты НА и слабого основания ВОН, не являющихся сопряженными в смысле протолитической теории Бренстеда - Лоури.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ РЕАКЦИИ В КАЧЕСТВЕННОМ АНАЛИЗЕ / ТЕМА 3.
Окислением наз-ют реакцию, связанную с потерей атомами или ионами одного или н-ких электронов, а восстановлением - реакцию, связанную с приобретением атомами или ионами электронов. Реакции ОВР взаимосвязаны и не могут рассматриваться изолированно друг от друга. Их называют еще редокс-реакциями. Окислителями называют вещества, принимающие электроны от другого вещества, а восстановителями - вещества, отдающие электроны другому веществу. Окислительно-восстановительная способность различных атомов и ионов характеризуется значением их нормального окислительно-восстановительного потенциала.
Его измеряют гальваническим элементом, состоящим из исследуемого металла, погруженного в раствор его соли и нормального водородного электрода. Потенциал нормального водородного электрода условно принят равным нулю при концентрации ионов водорода, равной 1н., и температуре 250С. Уравнение Нернста: значение ОВП зависит от температуры и концентрации ионов, присутствующих в растворе и выражается уравнением Нернста - для реакции
аА +bB=dD + eE; имеет вид E =E0+RT/nF ln[A]a[B]b/[D]d[E]e,
где Е - окислительно-восстановительный потенциал; Е0 -нормальный окислительно-восстановительный потенциал; n - число принимаемых или отдаваемых ионом электронов; R - молярная газовая постоянная, равная 8,314 Дж/(моль К); a,b,d,e -стехиометрические коэффициенты уравнения реакции; [A],[B],[D],[E] - концентрации ионов, моль/л; F - постоянная Фарадея, равная 96500 Кл; T -абсолютная температура, К. После подстановки в уравнение Нернста значений R, F, T = 298 K и перехода от натуральных логарифмов к десятичным получают выражение E=E0 +0,059/n lg [A]a[B]b/[D]d[E]e.
По этой формуле можно, в частности, вычислить значения потенциалов в различные моменты титрования, в области эквивалентной точки титрования вычисления производят по формуле
E =aE'0 + bE»0 / a + b, где E'0 и E»0
- стандартные окислительно-восстановительные потенциалы окислителя и восстановителя; a, b - число отдаваемых и принимаемых электронов.
Пример 1. Раствор содержит анионы Cl- и SO32-.
Какой из них будет окисляться перманганатом калия, если концентрации анионов равны? В соответствии с табличными данными нормальный ОВП для С1-→ С10 Е0 = 1,359 В, а для SO32- → SO42- E0 = 0,17 В. Разность стандартных потенциалов ионов, участвующих в реакции, называется электродвижущей силой реакции (ЭДС) реакции. Чем больше ЭДС, тем энергичнее протекает реакция. Для реакции окисления сульфит-иона ЭДС равна 1,51-0,17 =1,34 В, а для окисления хлорид иона 1,51-1,359 = 0,151В.
Следовательно, более энергично будет протекать реакция окисления сульфит-иона и только после его полного окисления начнет окисляться хлорид-ион.
Пример 2. Вычислить потенциал при титровании 100мл 0,1 н. раствора FeSO4 в точке, когда добавлено 99,9 мл 0,1 н. раствора KMnO4.Т.к. к 100мл раствора FeSO4 добавлено 99,9 мл раствора KMnO4, в растворе остается 0,1мл FeSO4 (100-99,9). Стандартный ОВП перехода Fe2+àFe3+ равен 0,771 В, число, отдаваемых при этом электронов п=1, т.о. Е=0,771+ 0,059/1 lg 99,9/0,1=0,771+0,059 lg 999=0,771+0,177=0,948 В.
Для сильных электролитов берут вместо концентраций активность. ЭДС равна разности между величиной потенциала окислителя и величиной потенциала восстановителя: ЭДС = Еокисл.-Евосст. Любая ОВР протекает при условии, если ЭДС реакции положительная. Упражнение: вычислите ОВП полуреакции Fe3+ + e-à Fe2+, если [Fe3+]=0,005 моль/л и [Fe2+] = 0,1 моль/л. Решение: по таблице находим редокс-пары E0 (Fe3+ / Fe2+) E = 0,771 + 0,0591 lg0,005 / 0,1= 0,0771 + 0,059·(lg(5·10-3)-lg10-1)= 0,771 + 0,059 -1,3 = 0,771-0,076 = 0,695 B. Степень окисления - это величина и знак заряда атома в соединении, рассчитанные таким образом, чтобы алгебраическая сумма всех зарядов в молекуле равнялась нулю, а в сложном ионе - заряду этого иона. Величина заряда определяется отношением электронов связи к наиболее электроотрицательному атому или делением электронов между 2-мя атомами.
Правила определения степени окисления:
- Степень окисления в простом веществе (т.е. в свободном состоянии) равна нулю.
- Щелочные металлы всегда имеют степень окисления +1, а щелочноземельные +2.
- Степень окисления фтора всегда равна -1.
- Степень окисления водорода равна +1 (кроме гидридов - его соединений со щелочными и щелочноземельными металлами, где она равна -1).
- .Степень окисления кислорода всегда равна -2 (кроме его соединений с фтором -F2O, где она равна +2, а также Н2О2, где она равна -1 и других пероксидов, производных от перекиси водорода).
Степени окисления - высшая, промежуточная и низшая.
Высшая положительная степень окисления характеризуется группой, в которой расположен данный элемент в таблице Д. И. Менделеева, например, Cl и Mn имеют высшую положительную степень окисления равную 7, т.к. располагаются в 7-ой группе. У азота высшая положительная степень окисления N равна 5, S имеет высшую положительную степень окисления равную 6-ти.
Степень окисления марганца в его соединениях MnO -+2 - низшая, Mn2O3 -+3 - промежуточная, MnO2-+4 -промежуточная, среда нейтральная, Mn3O4-+8/3 - промежуточная, среда щелочная, K2MnO4 -+6 -промежуточная, KMnO4 -+7 - высшая. НС1 -1, С12-0, НС1О -+1-низшая, НС1О2-+3 - промежуточная, НС1О3 -+5 - промежуточная, НС1О4-+7 - высшая. Атом элемента в своей высшей степени окисления не может ее повысить (отдать электроны) и поэтому проявляет только окислительные свойства, а в своей низшей степени окисления не может ее понизить (принять электроны) и проявляет только восстановительные свойства. Атом элемента, имеющий промежуточную степень окисления, может проявлять как окислительные, так и восстановительные свойства. При ОВР валентность атомов может и не меняться. Например, Н02 + С102 = 2Н+С1- валентность атомов водорода и хлора до и после реакции равна единице, т.к. валентность определяет число связей, образованных данным атомом, и поэтому знака заряда не имеет. А степень окисления атомов водорода и хлора изменилась и приобрела знаки + и -.
Все металлы проявляют только восстановительные свойства. Если металлическую пластинку опустить в воду, то катионы металла на ее поверхности гидратируются полярными молекулами воды и переходят в жидкость. При этом электроны, в избытке остающиеся в металле, разряжают его поверхностный слой отрицательно. Возникает электростатическое притяжение между перешедшими в раствор гидратированными катионами и поверхностью металла. В системе устанавливается подвижное равновесие: Me + m H2O↔Me (H2O)n+m (в растворе) + ne- (на металле), где n - число электронов, принимающих участие в процессе.
На границе металл - жидкость возникает двойной электрический слой, характеризующийся определенным скачком потенциала - электродным потенциалом. Абсолютные значения электродных потенциалов измерить не удается, т.к. они зависят от многих факторов и оперируют обычно относительными электродными потенциалами при определенных условиях - их называют стандартными электродными потенциалами. Стандартным электродным потенциалом металла называют его электродный потенциал, возникающий при погружении металла в раствор его соли (или собственного иона) с концентрацией (или активностью), равной 1моль/л, и измеренному по сравнению со стандартным водородным электродом, потенциал которого при 250С условно принимается равным нулю (Е0=О,ΔG0=0 - это изобарно-изотермический потенциал или энергия Гиббса.) Мерой химического сродства является убыль энергии Гиббса, которая зависит от природы вещества, его количества и температуры и является функцией состояния.
ΔGх.р. =∑ΔGпрод. - ∑ΔGисх.обр..
Самопроизвольно протекающие процессы идут в сторону уменьшения потенциала и, в частности, в сторону уменьшения ΔG и если ΔG<0, процесс принципиально осуществим; еслиΔG>0, процесс самопроизвольно проходить не может. Чем меньше ΔG, тем сильнее стремление к протеканию данного процесса и тем дальше он от состояния равновесия, при котором ΔG=0 и ΔH = TΔS). Располагая металлы в ряд по мере возрастания их стандартных электродных потенциалов (Е0), получаем ряд стандартных электродных потенциалов (ряд напряжений). Положение металла в ряду напряжений характеризует его восстановительную способность, а также окислительные свойства его ионов в водных растворах при стандартных условиях. Чем меньше значение Е0, тем большими восстановительными свойствами обладает данный металл в виде простого вещества и тем меньшими окислительными способностями характеризуются его ионы и наоборот. Электродные потенциалы измеряют приборами - гальваническими элементами. ОВР гальванического элемента протекает в направлении, в котором ЭДС элемента имеет положительное значение. В этом случае ΔG0 < 0, т.к. ΔG0 = -nFE0.
Электродный потенциал металла (Е) зависит от концентрации его ионов в растворе. Решение задач об изменении значений электродных потенциалов при различных концентрациях их ионов в растворе решается с помощью уравнения Нернста Е = Е0 + 0,059/n·lgc, где с - концентрация (при точных вычислениях активность) гидратированных ионов металла в растворе, моль/л. Соответственно, зная электродный потенциал металла с помощью уравнения Нернста можно определить концентрацию ионов металла в растворе в моль/л. Также с помощью уравнения ЭДС можно определить какой металл будет в гальваническом элементе катодом, а какой анодом (с меньшим потенциалом - это анод). Е = Е0 - в формуле Нернста при условии, что [ох] =[rеd]=1моль/л. Значение Е0 различных ОВ систем приводится в справочных таблицах.
Т. например, Е(Fе3+/Fе2+) = Е0(Fе3+/Fе2+) + 0,059 lg ([Fе3+]/[Fе2+]); Е(Fе3+/Fе2+)=0,77+0,059 lg ([Fе3+]/[Fе2+]).
Значение редокс-потенциала определяют по отношению к стандартному водородному электроду, потенциал которого принят за 0.
КОМПЛЕКСООБРАЗОВАНИЕ В КАЧЕСТВЕННОМ АНАЛИЗЕ, ковалентная связь, донорно-акцепторный механизм, тема 4
Комплексными называют соединения, в которых хотя бы одна ковалентная связь образована по донорно-акцепторному механизму, т. е. с помощью ковалентной связи, при образовании которой на связывающую МО (молекулярную орбиту) переходят два электрона лишь одного из партнеров, так что новой электронной пары не образуется. Например, при образовании иона аммония NH4+ из молекулы аммиака и иона НГ на образующуюся четвертую связывающую МО переходят электроны только аммиака (пример на электронной схеме, стр. 50, Новый справочник по химии для школьников и абитуриентов / Гузей Л.С, Кузнецов В.Н. Под редакцией проф. С.В. Дунаева. - 1-е изд. - М.: Большая медведица, 1998 - 354 с).
Поскольку подобное описание удобно при рассмотрении многих комплексных (координационных) соединений, донорно-акцепторную связь нередко называют также координационной. По физической природе донорно-акцепторная связь не отличается от обычной ковалентной связи, особенно если рассматривать образование соответствующих молекул непосредственно из атомов. Способность образовывать комплексные соединения наиболее сильно выражена у d-эдементов больших периодов периодической системы. Естественно при их анализе комплексообразование должно играть важную роль.
Как известно все d-элементы это металлы (железо, цинк, марганец, медь) с широким спектром химических свойств и их определение осуществляется с помощью гексцианоферрата калия.
В соответствии с координационной теорией Вернера центральный ион молекулы комплексного соединения, несущий обычно положительный заряд называется комплексообразователем. Он удерживает (координирует) в непосредственной близости некоторое число ионов с противоположным зарядом (или полярных молекул), называемых лигандами (или аддендами). Ион-комплексообразователь и лиганды вместе составляют внутреннюю координационную сферу.
Важнейшие лиганды - это нейтральные, но полярные молекулы
(Н2О, NH3, NO, СО и др.), а также многие анионы (СГ, Br ", NO2\ CN, ОН, S2O3 "2, Г).
Лиганды - монодентантные (NH3, метиленамин - CH3NH2, NO2", CN"), занимающие одно место во внутренней координационной сфере, и полидентантные (СО3", SO4, S2O3") - два и более мест.
Двухзарядный анион этилендиаминтетрауксусной кислоты (ЭДТА) - пример 4-хдентантного лиганда.
Следовательно, дентантность определяется числом атомов в лиганде, которыми он связан с комплексообразователем. Число ионов или полярных молекул, координируемое комплексообразователем во внутренней сфере, называют молекулами-отрицательными» - ионами. Эти" лигандев (донорных групп), координированных центральным атомом (акцептором), и образуют внутреннюю координационную сферу. При изображении формул комплексных соединений комплекс, составляющий внутреннюю координационную сферу, изображается в квадратных скобках.
Т.о. структура комплексного соединения представлена координационной (внутренней) сферой, состоящей из центральной частицы - комплексообразователя (ион или атом) - и окружающих ее лигандов (ионы противоположного знака или молекулы). Т.о. внутренняя сфера (комплекс) может быть анионом, катионом и не иметь заряда.
Например, в комплексном соединении
K3[Fe(CN)6],
внешняя сфера ЗК, внутренняя сфера [Fe(CN)6], где Fe3+ -комплексообразователь, a 6CN - лиганды, причем 6 - координационное число. Максимальное число лигандов, которое способен удерживать центральный ион, наз-ся координационным числом. Ионы, находящиеся за пределами координационной сферы, образуют внешнюю сферу комплексного соединения. Т.о. комплексное соединение (как правило) в узлах кристаллической решетки содержит комплекс, способный к самостоятельному существованию в растворе.
А. Вернер систематически изложил свою теорию и экспериментальный материал, подтверждающий ее положения и выводы в работе «Новые воззрения в области неорганической химии» в 1905 г. Он показал, в частности, что платина (IV), кобальт (111), иридий (111) и хром обладают координационным числом 6, а платина (11), палладий (11), медь (11) проявляют 4-е побочных валентности (т.е. координационное число равно 4). Известны комплексообразователи с координационными числами 2, 3, 7 и 8. Главные валентности комплексообразователя насыщаются только отрицательными ионами. В тоже время побочные валентности могут насыщаться как отрицательными ионами, так и нейтральными молекулами.
Этим и объясняется, что в соединениях, содержащих хлор, только ионы хлора могут осаждаться нитратом серебра, атомы же хлора, непосредственно связанные с комплексообразователем, не реагируют с нитратом серебра, например,
[Сг(Н2О)2(№1з)зС1]С12.
А. Вернер пользуясь определениями электропроводности, установил число ионов в соединениях и тем самым состав внутренней координационной сферы. Для примера определим заряд, координационное число и степень окисления комплексообразователя в комплексной соли K4[Fe(CN)6]. Заряд комплексного иона равен заряду внешней сферы, но противоположен ему по знаку. Координационное число комплексообразователя равно числу лигандов, координированных вокруг него, т.е. 6. Степень окисления комплексобразователя определяется также, как степень окисления атома в любом соединении, исходя из того, что сумма степеней окисления всех атомов в молекуле равна нулю. Заряды нейтральных молекул (Н2О, NH3) равны нулю.
Заряды кислотных остатков определяют из формул соответствующих к-т, т.о. степень окисления комплексообразователя +2. Многие соли, также как и гексацианферрат калия, являясь сильным электролитом, в водном р-ре необратимо диссоциируют на ионы внешней и внутренней сфер K4[Fe (С1Ч)б] = 4К7+[Те(СК)бГ\ т0 комплексный ион диссоциирует обратимо и в незначительной степени на составляющие его частицы: [Fe (CN)6]4~<-»Fe2++6CN~ Обратимый процесс характеризуется своей константой равновесия, которая в данном случае называется константой нестойкости (KJ комплекса.
KH=[Fe+] [CN"]7 [[Fe(CN)6]'"].
Чем меньше Кн тем более прочен данный комплекс Как правило, координационное число атома металла не совпадает с его степенью окисления: обычно координационное число атома металла выше его степени окисления. Число координационных связей, образуемых одним и тем же лигандом с одним атомом металла-комплексообразователя, наз-ся дентантностъю (старое наз-ие координационная емкость). Т.о. лиганды могут быть монодентантными и полидентантными (би, три, тетра, пента, гексадентантными). Монодентантные лиганды - это анионы F, СГ, Вг", Г, Н", CN", NO"2, SCN", нейтральные молекулы, имеющие только один донорный атом - аммиак NH3, первичные амины R - NH2, молекулы воды. Они обычно образуют одну координационную связь (кроме образования мостиковых связей между 2-мя атомами металла), стр. 184, 1-ый том, Харитонов. Координационная связь металл-лиганд может быть полярной ковалентной, по происхождению донорно-акцепторной, так в тетрааммиачном комплексе меди(11)[Си(№1з)4]2+, донорно-акцепторная связь Си11-*-NH3.
Каждая такая координационная связь медь(11)-аммиак образуется за счет оттягивания «свободной» электронной пары от молекулы аммиака на пустую орбиталь меди(П), что в записи химической формулы обозначается стрелкой, направленной от донора к акцептору. Наиболее распространенными являются три подхода к пониманию природы химической связи в координационных соединениях металлов: теория кристаллического поля, метод валентных связей и теория молекулярных орбиталей. Комплексные соединения могут быть катионного, анионного типа и комплексами-неэлектролитами, точнее слабыми электролитами. Внутренняя сфера комплексов катионного типа несет положительный заряд, например, [Cu(NH3)4]+,[Ag(NH3)2], [Co(NCS)2En2] -En - молекула этилендиамина. Внутренняя сфера комплексов анионного типа несет отрицательный заряд, например, [Ag(S2O3)2]J~, [Sb(OH)6]+, [Со(Ж>2)б]"". Внутренняя сфера комплексов-неэлектролитов не несет никакого электрического заряда, например, [R(NH3)2C12]. He менее важными являются выводы А. Вернера, касающиеся стереохимии комплексных соединений. Основное положение состоит в том, что молекулы и ионы, связанные с центральным ионом побочными валентностями, располагаются вокруг него в пространстве как в кристаллическом состоянии, так и в растворах. Пространственное строение внутренней сферы комплексов может быть линейным, треугольным, квадратным, тетраэдрическим и т.д. в зависимости от природы центрального атома металла, лигандов, внешнесферного окружения. Комплексные соединения катионного и анионного типов чаще всего растворимы в воде, комплексы-неэлектролиты, как правило, малорастворимы в воде. Вначале, как уже указывалось выше на примере ферроцианида калия происходит первичная диссоциация - отщепляются ионы внешней среды. При первичной диссоциации комплекса, имеющего ионы во внешней сфере, соединение ведет себя как сильный электролит. Затем происходит вторичная диссоциация уже по типу слабого электролита - отщепляются лиганды внутренней сферы.
Ступень диссоциации внутренней сферы комплекса
Каждая ступень диссоциации внутренней сферы комплекса характеризуется своей константой химического равновесия, формулы стр. 191.
- Внутрикомплексные (хелатные) соединения.
- Биокоординационные соединения (хелаты);
- Применение хелатов в аналитической химии;
- Роль биокоординационных соединений в живых объектах.
. Интересным особым классом комплексов являются так называемые внутрикомплексные соединения (иначе хелаты), в которых комплексообразователь одновременно связан с двумя или более атомами одного и того же лиганда (полидентантные лиганды) [1]. Простейшим примером может служить гликоколят меди [(NH2CH2COO)Cu], в котором каждый аминоацетат-анион присоединен к Си2+ валентной связью через кислород и донорной через азот. Центральный атом оказывается тем самым как бы втянутым внутрь лиганда (от чего соединения такого типа и получило название внутрикомплексным). Такая функция лигандов характерна, в частности, для «трилонов» (см. ниже).
Как правило, типичные хелаты лучше растворимы в органических растворителях, чем в воде. Их водные растворы показывают ничтожную электропроводность. По отношению к различным внутрикомплексные соединения большей частью весьма Например, из раствора гликоколята меди последняя не сероводородом.
. Образование хелатов часто используется в аналитической химии, для маскировки катионов при определении микроколичеств элементов в различных биологических объектах.
- Полидентантные лиганды образуют различные циклы, например:
- реактивам устойчивы, осаждается кислота
- Никель с диметилглеоксимом образует два цикла.
- Комплекс Ti с хроматхолевой кислотой содержит 6 циклов.
- Строение и устойчивость бионеорганических соединений с полидентантными лигандами зависит от величины цикла и его сопряженности].
Отдельно рассмотрим полидентантные лиганды в этилендиаминотетрауксусной кислоте. С такими катионами как Са + и Mg + лиганды образуют четырех членные циклы, а также они склонны к образованию двух ядерных комплексов, образованию комплексов с дополнительной связью Me - Me. Такие комплексы применяются для количественного определения Са2+, Mg2+ в воде.
Фосфоглицериновая кислота присоединяет вторую фосфатную группу от аденозинтрифосфата (АТФ), а дифосфоглицериновая кислота восстанавливается с помощью НАД-Н в фосфотриозу, участвующую в синтезе Сахаров от Сз до С7. Продуктами этой стадии являются АТФ и фруктозофосфат, превращающийся в дальнейшем в глюкозу и крахмал.
Гемоглобин - гемсодержащий белок, обратимо связывающий молекулярный кислород (рис. 13.7):
нь + о2-ньо2
Из рис. 13.8 видно, что как и Mg2+ в хлорофилле, Fe2+ связан с четырьмя атомами азота пиррольных колец порфириновой системы (протопорфирина IX) и с атомами азота имидазольного кольца остатка аминокислоты - гистидина, входящего в состав полипептидной части гемоглобина. Шестое координационное место занимает дикислород или другие малые лиганды (СО в карбоксигемоглобине НЬСО).
Молекула гемоглобина
НЬСО образуется в сотни раз быстрее, чем НЬО2, чем и объясняется токсичное действие СО. Еще более токсичен NO. Роль белковой части
3. К числу соединений этого типа относятся такие важные для жизни вещества, как хлорофилл и гемоглобин. Со структурной точки зрения оба эти катализатора жизненных процессов сходны друг с другом.
Биокоординационные соединения выполняют важнейшие функции в растительных и животных организмах. Они в составе ферментов катализируют реакции переноса кислорода, окислительно-восстановительные и гидролитические процессы, чаще всего биокоординационные соединения представляют собой металлсодержащие макромолекулы.
Связанный с белком металл может контролировать конформацию биомакромолекул. Так, Mg2+, Ca + стабилизируют двойную спираль ДНК, тогда как Си2+ благоприятствует ее раскручиванию. В этом параграфе мы ограничимся некоторыми примерами биокоординационных соединений, ответственных, в частности, за фотосинтез, перенос кислорода, окислительно-восстановительные процессы.
Хлорофилл. Этим термином определяют группу магнийсодержащих пигментов, ответственных за процессы фотосинтеза. Наиболее распространенным является хлорофилл а, который содержится во всех растениях, образующих кислород в процессе фотосинтеза. Кроме него известны не менее восьми разновидностей.
В процессе фотосинтеза солнечная энергия поглощается хлорофиллом. Высвобождаемый кислород первоначально принадлежал воде. Водород восстанавливает фермент никотинамидадениндинуклеотид (НАД), в результате чего образуются НАД-Н и Н+, которые принимают участие в цикле синтеза углеводов. Эта стадия называется световой.
БУФЕРНЫЕ РАСТВОРЫ, БУФЕРНАЯ ЕМКОСТЬ, электролит, электролитическая диссоциация воды ТЕМА 5
По уравнению электролитической диссоциации воды видно, что вода типичный электролит, проявляющий амфотерные свойства: Н2О <-> 2Н+ + ОН- или Н2О + Н2О <-> Н3О+ + ОН-. Однако, для простоты вместо иона гидроксония, в виде которого водород находится в растворе, пишут и говорят об ионе водорода (такая замена не влияет на расчет). Т. о. при диссоциации воды образуются ионы водорода, понимаемые как ионы гидроксония - носители кислотных свойств и ионы гидроксила - обуславливающие щелочные свойства воды. Как видно из уравнения диссоциации в жидкой воде концентрация ионов водорода приблизительно равна концентрации ионов гидроксила и поэтому можно написать, что в воде [Н+] = [ОН-]. Применив закон действующих масс к равновесной системе электролитической диссоциации воды получим:
[Н+][ОН-] / [Н2О] = К, т. е. получаем константу электролитической диссоциации воды.
Подобные процессы называют реакциями автопротолиза, в этих реакциях одна молекула амфипротного растворителя ведет себя как кислота, а другая как основание. Вычисления по электрической проводимости показали, что концентрация ионов водорода и гидроксид-ионов в воде оказалась равна 10-7 моль/л при 220 С. Преобразуя это уравнение, получаем [Н+][ОН-] = [Н2О] К. Но степень диссоциации воды очень мала, и концентрацию ее недиссоциированных молекул в любом разбавленном водном растворе можно считать величиной постоянной. Т. о. в правой части уравнения находятся 2-ве постоянные величины: [Н2О] - концентрация недиссоциированных молекул воды и К - константа диссоциации. Но произведение 2-х постоянных величин есть также величина постоянная.
Поэтому заменив [Н2О]К новой константой, получим
[Н+] [ОН-] = КН2О.
Следовательно, как бы не изменялись концентрации ионов Н+ и ОН- в воде или в разбавленном водном растворе, произведение их остается величиной практически постоянной. Эту величину называют ионным произведением воды. Числовое значение этой константы нетрудно найти, подставив в уравнение значения концентраций водородных и гидроксид-ионов в воде:
КН2О = [Н+][ОН-] = 10-710-7 = 10-14.
Реакцию того или иного раствора принято характеризовать концентрацией водородных ионов, т.к. концентрацию ионов ОН- легко вычислить, исходя из ионного произведения воды. Концентрация ионов Н+ и ОН- изменяется в обратно пропорциональной зависимости, но никогда не становится равной нулю:
Н+= КН2О / ОН- или ОН- = КН2О/Н+.
Т. к. ионное произведение воды при данной температуре неизменно и равно 10-14, а величины [Н+] и [ОН-] переменны, то по этим их величинам можно судить о кислотности или щелочности раствора. Реакция среды кислая, если величина [ОН-] уменьшается, т. е. становится меньше 10-7 г-ион/л; реакция щелочная при соответственном увеличении концентрации ионов ОН-.
Следовательно, всякий раствор будет называться нейтральным, если в нем при комнатной температуре [Н+] = [ОН-] = 10-7 г-ион/л. Для кислых растворов справедливо условие
[Н+]>[ОН-]; [Н+]>10-7 г-ион/л, [ОН-]<10-7.
Для щелочных растворов справедливо
- [Н+]<[ОН-]; [Н+]<10-7, [ОН]<10-7 г-ион/л.
Например, если концентрация ионов водорода в растворе равна 10-3 г-ион/л, реакция среды кислая, величину [ОН-] в таком растворе можно рассчитать из уравнения ионного произведения воды:
[Н+][ОН-] = 10-14г-ион/л, следовательно, [ОН-] =10-14/10-3=10-11 г-ион/л.
Ионизация воды эндотермический процесс, протекающий с поглощением тепла, согласно закону Ле-Шателье при повышении температуры равновесие ионизации смещается в сторону образования ионов, что приводит к увеличению ионного произведения воды. Зная экспериментальные табличные данные ионного произведения воды можно решать различные задачи. Однако, характеризовать основность и кислотность растворов концентрацией ионов водорода с отрицательным показателем степени оказалось практически неудобным. И поэтому в практике применяют водородный показатель - рН, отрицательный логарифм концентрации ионов водорода: р
Н= -lg [Н+]. Т. о. нейтральная среда рН=7, кислая рН<7, щелочная рН>7.
Чем меньше величина рН, тем больше концентрация водородных ионов, тем больше кислотность раствора. Кислотность раствора растет с уменьшением рН, в то время как щелочность возрастает с его увеличением. Все сказанное наглядно видно, если изобразить значения рН в виде схемы.
Конечные точки этой схемы соответствуют, примерно, концентрации ионов водорода 1н. раствора НС1 (рН=0) и 1н. раствора NаОН (рН=14). Конечно могут быть более кислые растворы с рН<0 и более щелочные с рН >14, но в этих случаях кислотность или щелочность этих растворов принято выражать в молях кислоты или щелочи на литр раствора. Наряду с водородным показателем нередко применяют гидроксильный показатель рОН= -lg [ОН-].
Если уравнение ионного произведения воды [Н+][ОН-] = 10-14 прологарифмировать, а затем поменять у логарифмов знаки на обратные, то получим
рН + рОН= рКН2О=14, где рКН2О= --lgКН2О=-lg10-14=14.
Отсюда следует, что в любом водном растворе действительно соотношение
рН= рКН2О - рОН = 14 - рОН (при 250 С).
Т. о., используя эти уравнения можно решать различные задачи: вычислять рН растворов кислот и оснований, вычислять концентрацию ионов Н+ и ОН- по известным значениям рН и обратно. При расчете водородного показателя среды водных растворов сильных кислот и оснований следует учитывать необратимость процесса их электролитической диссоциации (α=1). Концентрация ионов Н+ в растворах одноосновных сильных кислот и концентрация ионов ОН- в растворах одноосновных щелочей численно равна молярности растворов. Например, концентрация ионов Н+ в 0,001М растворе НС1 равна 0,001 моль/л, а концентрация ОН- в 0,01 М растворе NаОН равна 0,01 моль/л. Для успешного проведения качественных реакций в нужном направлении необходимо соблюдение определенных условий, в первую очередь, определенной величины рН исследуемого раствора. Если требуемая величина рН не соблюдается, как правило, реакция не идет в нужном направлении и всю работу приходится начинать с начала.
Так, например, катион магния отнесен к 1-ой группе катионов (по кислотно-основной схеме определения катионов) и его можно осадить из раствора, содержащего ион магния в виде гидроокиси, согласно уравнению Мg2++2ОН--->Мg(ОН)2. Однако эта реакция идет только при определенном значении рОН-<14, т. е. при рН >10. При добавлении к раствору соли магния гидроокиси аммония осадок Мg(ОН)2 не выпадает даже при рН=9 или осаждение бывает неполным. Полное осаждение происходит при действии сильных оснований, а в присутствии солей аммония гидрат окиси магния Мg(ОН)2 остается в растворе.
Поэтому магний относят к первой группе катионов. Учитывая вышеизложенное, при проведении качественных реакций необходимо указывать не только характер среды: «кислая» либо «щелочная», но и точное значение рН. Методы определения рН среды: колориметрический и потенциометрический.
Потенциометр, иономер измерение рН с помощью электролитической ячейки и двух электродов
Наиболее точный потенциометрический метод, который осуществляется с помощью прибора потенциометра или иономера. Прибор необходимо предварительно подготовить и произвести настройку с помощью тест-стандартов или нормодоз, т. е. растворов с известным значением рН, измерение рН осуществляется с помощью электролитической ячейки и двух электродов. Колориметрический метод основан на применении реактивов, изменяющих свою окраску в зависимости от концентрации водородных ионов, которые называются индикаторами Это слабые органические кислоты или основания недиссоциированные молекулы которых имеют различную окраску, они называются кислотно-основными индикаторами. Лакмус, например, содержит азолитминовую кислоту недиссоциированные молекулы которой красного цвета, а анионы синего. Добавление к раствору кислоты или щелочи изменяет рН раствора с чем и связано изменение окраски, рассматриваемых индикаторов.
Важным условием соблюдение значений рН является при определении амфотерных катионов 4-ой группы.
- lg [Н+]- -lg [ОН-]= lgКН2О = 14.
Для приблизительного определения рН среды в ходе качественного анализа чаще всего пользуются следующими индикаторами: метиловым оранжевым, метиловым красным, лакмусом, фенолфталеином, феноловым красным, малахитовым зеленым и др. К отдельным пробам испытуемого раствора добавляют по 1 - 2 капли раствора каждого индикатора. При этом нужно знать какую окраску имеет используемый индикатор в зависимости от рН среды. Например лакмус (или лакмусовая бумажка) краснеет в кислой среде при рН <5, а синеет в щелочной среде при рН>8. Для фенолфталеина малиновая окраска характерна в щелочной среде (при рН>10; начиная от рН 8,2 и меньше он бесцветен. Универсальные индикаторы, например, ЗИВ-1, представляет собой сухую смесь из 5-и индикаторов: диметиламиназобензола, бромтимолового синего, метилового красного, фенолфталеина и тимолфталеина. Готовая смесь в ампулах, в количестве 0,16 г растворяется в 100 мл 80 %-го спирта. В зависимости от рН раствора универсальный индикатор приобретает окраску в соответствии со шкалой. Действие одноименных ионов.
Степень электролитической диссоциации вещества зависит не только от его концентрации в растворе, но и от добавления в раствор других электролитов. Степень диссоциации слабого электролита можно понизить, добавляя в раствор сильный электролит, содержащий ион, одноименный с первым электролитом. Это явление объясняется с точки зрения закона действующих масс и константы равновесия реакции диссоциации. Предлагается самим студентам объяснить изменение степени диссоциации раствора уксусной кислоты при добавлении уксуснокислого аммония или натрия и раствора аммиака при добавлении раствора хлорида аммония. Т. о. степень диссоциации слабого электролита понижается при введении в раствор какого-нибудь сильного электролита содержащего одноименный ион. Растворы, рН которых почти не изменяется от прибавления небольших объемов сильных кислот или щелочей, а также от разбавления называют буферными растворами или буферными смесями. Они представляют собой смеси электролитов, содержащие одноименные ионы.
Например, ацетатный буферный раствор - это смесь уксусной кислоты и ацетата натрия, аммонийный буферный раствор - смесь гидрата окиси аммония и хлористого аммония. Ацетатная смесь проявляет свое буферное действие только до тех пор, пока концентрация прибавленной кислоты или щелочи не превысит приблизительно 0,08 моль/л. Количество молей сильной кислоты или сильного основания, прибавление которого к 1 л буферного раствора изменяет его рН на единицу, характеризует буферную емкость раствора. Следовательно, буферная емкость измеряется количеством г-экв или моль/л сильной кислоты или сильного основания, которое необходимо добавить к 1 л буферной системы, чтобы изменить его значение рН на единицу. Буферным действием обладают также смеси кислых солей с различной замещенностью водорода металлом.
Натрия первая соль играет роль слабой кислоты, а вторая - роль её соли
В буферной смеси дигидрофосфата и гидрофосфата натрия первая соль играет роль слабой кислота, а вторая - роль её соли.
Фосфатный буферный раствор
(NаН2РО4 + NаНРО4) поддерживает рН 6,8.
Т. о. варьируя концентрации слабой кислоты и её соли, можно получить буферные растворы с разными значениями рН.
Примерами буферных действий в природе является кровь, лимфа, почва. Для вычисления рН в буферных смесях применяют уравнение рН = рКк - lg(ск/сс), где рК является силовым показателем кислоты или это логарифм константы диссоциации кислоты; ск - концентрация кислоты, а сс - концентрация соли. Указанное уравнение получено после логарифмирования уравнения константы диссоциации слабой кислоты, входящей в буферную смесь. Для смесей слабых оснований и их солей рОН = рКо - lg(со/сс).
Учитывая, что
рН = рОН = 14,
можно написать
рН = 14 - рОН = 14 - рКо + lg(со/сс). Т
.о. 1. мах буферная емкость набл-ся у смесей содержащих равные концентрации компонентов;
2. По мере добавления к буферному раствору кислоты или щелочи устойчивость его к изменению рН постепенно уменьшается. 3. Буферная емкость раствора тем больше, чем выше концентрация компонентов буферной смеси. При проведении анализов при постоянном рН следует учитывать буферную емкость используемой смеси.
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ, ЗАДАЧИ И МЕТОДЫ. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ, химические, физические и физико-химические методы / ТЕМА 6
Методы количественного анализа. Количественный анализ предназначен для определения количественного состава анализируемого вещества. Существуют химические, физические и физико-химические методы количественного анализа. Основой всякого количественного исследования является измерение. Химические методы количественного анализа основаны на измерении массы и объема.
Количественные исследования позволили ученым установить такие основные законы химии, как закон сохранения массы вещества, закон постоянства состава, закон эквивалентов и др. законы, на которых основана химическая наука. Принципы количественного анализа являются основной для химико-аналитического контроля производственных процессов различных отраслей промышленности и составляют предмет т. н. технического анализа. Различают 2-ва основных метода количественного химического анализа: весовой или гравиметрический и объемный или титриметрический.
Весовым анализом наз-ся метод количественного анализа, в котором точно измеряют только массу. Объемный анализ - основан на точном измерении массы веществ и объема раствора реактива известной концентрации, реагирующего с определенным количеством анализируемого вещества. Особым видом кол-го анализа является анализ газов и газовых смесей, т.н. газовый анализ, выполняемый тоже путем измерения объема или массы анализируемой смеси или газа.
Определение одного и того же вещества можно выполнить весовым или объемным методами анализа. Выбирая метод определения, аналитик должен учитывать необходимую точность результата, чувствительность реакции и быстроту выполнения анализа, а в случае массовых определений - доступность и стоимость применяемых реактивов.
В связи с зтим различают макро-, микро-, полумикро-, ультрамикрометоды кол-го анализа, при помощи которых можно проводить анализ минимальных количеств анализируемого вещества.
Простые химические методы все больше вытесняются физическими и физико-химическим методами,
для работы с которыми необходимы дорогостоящие приборы и оборудование.
- Оптические,
- электрохимические,
- хроматографические,
- различные спектро- и фотометрические исследования (инфракрасная, атомноадсорбционная, пламенная и т.д.),
- потенциометрия,
- полярография,
- масс-спектрометрия,
- ЯМР исследования.
Данные методы ускоряют получение результатов, повышают их точность и чувствительность измерений: предел обнаружения (1-10-9мкг) и предельная концентрация (до 10-15 г/мл), селективность (можно определять составные компоненты смеси без их разделения и выделения), возможность их компьютеризации и автоматизации.
Но с другой стороны все более удаляют от химии, снижают знание химических методов анализа у аналитиков, что и привело к ухудшению преподавания химии в школах, отсутствию хороших учителей-химиков, оснащенных школьных химических лабораторий, снижению знаний по химии у школьников. К недостаткам следует отнести сравнительно большую ошибку определения (от 5 до 20 %, в то время как химический анализ дает ошибку обычно от 0,1 до 0,5 %), сложность аппаратуры и ее высокую стоимость.
Требования, предъявляемые к реакциям в количественном анализе. Реакции должны протекать быстро, до конца, по возможности, - при комнатной температуре. Исходные вещества, вступающие в реакцию, должны реагировать в строго определенных количественных соотношениях (стехиометрически) и без побочных процессов. Примеси не должны мешать проведению количественного анализа. При проведении измерений не исключены ошибки, погрешности измерений и расчетов. Для исключения ошибок, сведения их к минимуму, измерение проводят в повторностях (параллельных определениях), не менее 2-х и проводят метрологическую оценку результатов (имеется в виду правильность и воспроизводимость результатов анализа).
Классификация химических методов количественного анализа:
Титриметрический метода. Измерение объема израсходованного на реакцию раствора реактива точно известной концентрации.
Гравиметрический. Измерение массы определяемого вещества или его составных частей, выделяемых в виде соответствующих соединений.
Важнейшими характеристиками методов анализа является их чувствительность и точность. Чувствительностью метода анализа называют наименьшее количество вещества, которое можно достоверно определить данным методом. Точностью анализа называют относительную ошибку определения, которая представляет собой отношение разности найденного (х1) и истинного (х) содержания вещества к истинному содержанию вещества и находят по формуле:
Отн. ош.= (х1-х)/ х, для выражения в процентах умножают на 100. За истинное содержание принимают среднеарифметическое содержание вещества, найденное при анализе пробы в 5 -7 определениях.
Весовым (гравиметрическим) анализом называют метод количественного анализа, при котором количественный состав анализируемого вещества устанавливают на основании измерений масс, путем точного взвешивания массы устойчивого конечного вещества известного состава, в которое полностью переведен данный определяемый компонент. Например, гравиметрическое определение серной к-ты в водном растворе осуществляется с помощью водного раствора соли бария:
ВаС12 + Н2SО4→ ВаSО4↓ +2 НСl.
Осаждение проводят в таких условиях, в которых практически весь сульфат-ион переходит в осадок ВаSО4 с наибольшей полнотой - количественно, с минимальными потерями, вследствие незначительной, но все же имеющейся растворимости сульфата бария.
Далее осадок отделяют от раствора, промывают для удаления растворимых примесей, высушивают, прокаливают, для удаления летучих сорбированных примесей и взвешивают на аналитических весах в виде чистого безводного сульфата бария. А затем рассчитывают массу серной кислоты. Классификация методов гравиметрического анализа. Методы осаждения, отгонки, выделения, термогравиметрические методы (термогравиметрия). Методы осаждения - определяемую составную часть количественно связывают в такое химическое соединение, в виде которого она может быть выделена и взвешена. Состав этого соединения должен быть строго определенным, т.е. точно выражаться химической формулой, и оно не должно содержать каких-либо посторонних примесей. Соединение, в виде которого определяемую составную часть взвешивают, называют весовой формой.
Пример, определение Н2SО4(выше), определение массовой доли железа в его растворимых солях, основанное на осаждении железа (111) в форме гидроксида Fе(ОН)3 хН2О с последующим его отделением и прокаливанием до оксида Fе2О3 (весовая форма). Методы отгонки. Определяемый компонент выделяют из анализируемой пробы в виде газообразного вещества и измеряют либо массу отогнанного вещества (прямой метод), либо массу остатка (косвенный метод). Прямой метод широко используется для определения содержания воды в анализируемых веществах путем ее отгонки из взвешенного образца и конденсации, а затем измеряют объем конденсированной воды в приемнике. По плотности пересчитывают объем воды на массу и, зная массу образца и воды, рассчитывают содержание воды в анализируемой пробе. Косвенный метод отгонки широко применяют для определения содержания летучих веществ (включая слабосвязанную воду) по изменению массы образца до и после высушивания до постоянного веса в термостате (в сушильном шкафу) при постоянной температуре.
Условия проведения таких испытаний (температура, время сушки) определяются природой образца и конкретно указываются в методических руководствах. Методы выделения основаны на выделении из раствора определяемого компонента путем электролиза на одном из электродов (электрогравиметрический метод). Затем электрод с выделевшимся веществом промывают, высушивают и взвешивают. По увеличению массы электрода с веществом находят массу выделившегося на электроде вещества (сплавы золота, меди переводят в раствор). Термогравиметрические методы не сопровождаются отделением исследуемого вещества, а исследуется сам образец поэтому эти методы условно относят к гравиметрическим методам анализа. Методы основаны на измерении массы анализируемого вещества при его непрерывном нагревании в заданном температурном интервале на специальных приборах - дериватографах.
По полученным термогравиграммам при их расшифровке можно определить содержание влаги и других составляющих анализируемого вещества. Основные этапы гравиметрического определения: расчет массы навески анализируемой пробы и объема (или массы) осадителя; взвешивание (взятие) навески образца; растворение навески анализируемого образца; осаждение, т.е. получение осаждаемой формы определяемого компонента; фильтрование (отделение осадка от маточного раствора); промывание осадка; высушивание и (при необходимости) прокаливание осадка до постоянной массы, т. е. получение гравиметрической формы; взвешивание гравиметрической формы; расчет результатов анализа, их статистическая обработка и представление.
Каждая из этих операций имеет свои особенности. При расчете оптимальной массы навески анализируемого вещества учитывают возможную массовую долю определяемого компонента в анализируемой пробе и в гравиметрической форме, массу гравиметрической формы, систематическую ошибку взвешивания на аналитических весах (обычно 0,0002), характер получаемого осадка - аморфный, мелкокристаллический, крупнокристаллический. Расчет исходной навески ведут исходя из того, что масса гравиметрической навески должна быть не меньше 0,1 г.
В общем случае нижний предел оптимальной массы m исходной навески анализируемого вещества (в граммах) рассчитывают по формуле:
= 100m (ГФ) F/ W(X),
где m(ГФ) - масса гравиметрической формы в граммах; F - гравиметрический фактор, фактор пересчета, аналитический множитель); W(X) - массовая доля (в %) определяемого компонента в анализируемом веществе. Гравиметрический фактор F численно равен массе определяемого компонента в граммах, соответствующий одному грамму гравиметрической формы.
Гравиметрический фактор рассчитывают по формуле как отношение молярной массы М(Х) определяемого компонента Х к молярной массе гравиметрической формы М(ГФ), умноженное на число n молей определяемого компонента, из которого получается один моль гравиметрической формы:
= n M(X) / M (ГФ).
Так, если из 2-х молей Fе С13 6Н2О получается один моль гравиметрической формы Fе2О3, то n = 2. Если из одного моля Ва(NО3)2 получают один моль гравиметрической формы ВаСrО4, то n = 1.
ГРАВИМЕТРИЧЕСКИЙ (ВЕСОВОЙ) МЕТОД АНАЛИЗА ТЕМА 7
Гравиметрический анализ. Аналитика строго говоря интересует не вес реагирующих веществв, а их массы. ( поскольку вес это непостоянный показатель и изменяется с изменением расстояния от тела до центра земли, на экваторе меньше, чем на полюсах, в невесомости равен нулю).
Мерой кол-ва материи (в обычных условиях) является масса, за ед. кот-й принят кг, ед-ей веса является ньютон. Т.о. атомный, молекулярный вес - это условные понятия, принятые в химии. Измерение массы при выполнении опред-ий методом весового анализа чаще всего должно быть очень точным и поэтому проводится обычно при помощи весьма точного прибора - аналитических весов.
Дать показатели точности при взвешивании на ан. весах. Выделяются 3-и типа весовых определений.
1 - весовые опред-ия при выполнении кот-ых опред-мую составную часть количественно выделяют из анализ-го в-ва и взвешивают. Количественно выделить - значит выделить составную часть анализируемого в-ва настолько полно, насколько это позволяют сделать лабораторные приемы и св-ва данного вещества.
Так определяют например сод-ие золы в каменном угле (это очень важный анализ, т. к. сод-ся в угле примеси снижают его теплотворные кач-ва и засоряют топку). Для проведения такого анализа берут навеску угля, т.е. точно взвешивают небольшое кол-во пробы угля и сжигают его в муфельной печи в тигле (тигель доведен до постоянного веса) и навеску прокаливают до тех пор, пока масса оставшейся золы не будет уменьшаться, тигель с остывшей в эксикаторе золой точно взвешивают и опред-ют зольность угля в процентах,
исходя из пропорции:
навеска - 100%,
масса золы - х%,
т.е. х= масса золы 100/навеска.
Т.о. методы выделения делятся на выделении в-ва путем сжигания, электролиза (далее стр. 3).
2. определяемую составную часть полностью удаляют, а остаток взвешивают.
Определение содержания влаги в исследуемых веществах.
Таким способом обычно определяют содержание влаги в исследуемых веществах. Привести пример и расчет определения влаги. Условия проведения таких испытаний на стр.3 в методе отгонки. Весовые опред-ия первых 2-х типов выполняются довольно просто, в 3-ем случае метод осаждения, когда невозможно, например выделить и взвесить серу из каменного угля применяется метод осаждения (стр.3).
В данном примере серу выделить в виде малорастворимого соединения ВаSО4. По кол-ву бария сернокислого легко опред-ть сод-ие серы, содержащейся в навеске угля, т. к. каждому атому серы соответствует одна мол-ла сер-го бария. Вычисления могут быть проведены при помощи соответствующих пропорций.
Для гравиметрических определений существует н-ко общих правил:
- Перед определением какой-либо составной части анализируемого в-ва из навески взятого образца должны быть тщательно удалены другие составные части, ведущие себя в процессе данного анализа также, как и определяемая, или приняты соответствующие меры, не допускающие выделения этих сопутствующих составных частей вместе с определяемой (при опред-ии серы вместе с барием могут выделяться сульфаты второй группы катионов).
- Следует использовать реактивы соответствующей степени чистоты: ч.д.а. или х.ч, обычно это указ-ся в методичках).
- Правильно выполнить расчеты, т.к. ошибка в расчете равноценна ошибке в анализе.
Средняя проба анализируемого в-ва - это небольшое кол-во этого в-ва, состав которого одинаков с составом всей партии, от которой взята проба. П
равила отбора средней пробы зависят от физического состояния продукта (твердое, жидкое, газообразное), структуры (крупнокусковое, мелкокусковое, зернистое, порошкообразное и т. п.), упаковки (банки, мешки, бочки, навалом, насыпом), способом перевозки (автомобили, железнод-ые вагоны, цистерны, баржи и т.д.), а также размером партии. Методы отбора проб: первичная большая из различных мест партии, квартование, разделение на 3-и равные части: на анализ, на повтор и на арбитражное хранение.
Все это определяется НТД на данный продукт. Навеска - небольшое, точно взвешенное кол-во анализируемого в-ва, взятое от средней его пробы, кот-ое в процессе анализа количественно подвергается всем необходимым операциям. Обычно это н-ко граммов или десятых грамма. От количества навески зависит точность результата анализа.
Об абсолютной и относительной ошибках мы говорили, ясно что чем больше навеска в-ва тем меньше будет относительная ошибка при одинаковой ошибке взвешивания (после 4-го знака после запятой). Но большая проба требует большего кол-ва реактивов, ее обработка требует большего времени (например сжигание, прокаливание).
При проведении гравиметрических опред-ий ведут предв-ый расчет навески.
Для весовых анализов 1 и 2-го типа навеска должна быть такой величины, чтобы выделяемая и удаляемая из навески определяемая часть составляла от 0,01 до 0,1 г. Следовательно, чтобы правильно рассчитать навеску нужно знать хим-ую формулу анализ-го в-ва или приблизительное сод-ие составной части в пробе.
Например, какую навеску образца каменного угля нужно взять для определения его зольности, если предпол-ая зольность 10%?. Т. к. золы в образце меньшее кол-во, то берем большую норму, т.е. 0,1г, чтобы навеска не получилась слишком маленькой и рассчитываем по пропорции: 0,1 -10%, х(навеска) - 100 %, получ-ся 1г. В третьем виде для образования осадка следует учитывать физические св-ва (структуру и плотность) в-ва, в виде которого осаждается определяемая составная часть, т.н. осаждаемая форма, которая часто отличается от весовой формы.
При определении железа осаждаемая форма Fе(ОН)3, а весовая форма - Fе2О3, в случае ВаSО4 и весовая и осаждаемая форма одинаковы, ионы Са осаждаются в виде СаС2О4Н2О, а взвешиваются после прокаливания осадка в виде СаО. Осаждаемая форма оксалат кальция, а весовая форма - окись кальция. Плотность получаемого осадка влияет на дальнейшие стадии его обработки, особенно на промывку - легко осадка будет много в фильтре и его трудно и долго придется промывать и в этом случае предпочтительнее тяжелый осадок.
Поэтому в осадительных методах величина навески должна рассчитываться исходя из характера осадка:
аморфного (легкого, гидроокиси) - 0,07 - 0,1г; кристаллического легкого (большинство солей) - 0,1 - 0,15г; кристаллического тяжелого - 0,2 - 0,4г; кристаллического очень тяжелого (соли свинца, серебра) до 0,5г.
Взятие навески на аналитических весах требует определенных навыков и выполняется по соответствующим правилам, переносят количественно в стакан или колбу смывая водой и растворяя в воде или к-те при нагревании в водяной или песочной бане и приступают к осаждению. Цель осаждения - количественно перевести определяемую составную часть анал-го в-ва в в опред-ое химич-ое соединение. Зная массу выделенного осадка, можно рассчитать сод-ие опред-ой составной части, если осаждение произойдет не количественно, то масса осадка получится меньшей, а след-но и результат анализа будет меньше.
И тут есть правило: если осаждаемый ион может быть осажден различными ионами, из них следует выбирать в кач-ве осаждающего тот, который образует осадок, обладающий наименьшей растворимостью. Для расчета кол-ва реактива необходимого для осаждения исходят из правила эквивалентности согласно которому 1г-экв осаждаемой составной части реагирует с 1 г-экв осадителя.
На практике осадителя берут в некотором избытке. При осаждении необходимо соблюдать следующие условия: осаждать только из разбавленных р-ов разбавленными растворами осадителя (концентрированные р-ры следует использовать при получении аморфных осадков). Этот прием позволяет медленно и количественно образовываться осадку и и затрудняет все виды соосаждения; осаждать только подогретые р-ры горячими растворами осадителей, что способствует образ-ию крупных кристаллов; осадитель в кол-ве вычисленного объема добавлять медленно, порциями, при постоянном помешивании стекл-ой палочкой; следует добавлять небольшое кол-во коагул-х в-в, желательно улетучив-ся при прокаливании осадка. Дать время для созревания осадка, т.е. для превращения мелких кристаллов в крупные. Аморфные осадки подвергают дальнейшей обработке сразу, без выдержки для созревания, т.к. они ведут себя в р-ре как адсорбенты, т. е. загрязняются.
Проверка полноты осаждения добавлением по стенке стакана н-ких капель осадителя и при появлении мути добавляют н-ко мл раствора осадителя и выдерживают некоторое время на водяной бане. Фильтрование и фильтрование с декантацией - сливание отстоявшейся жидкости по стеклянной палочке, стараясь не взмучить р-р. Используют воронки стеклянные или другие, фильтр складывают, раскрывают, вставляют в воронку, расправляют, смачивают дист-ой водой, между стенками воронки и фильтром не должно оставаться пузырьков воздуха. Затем добавляют 50 мл промывной жидкости взмучивают, дают отстояться и вновь декантируют и так трижды, практически большее количество промывок, что обеспечивает лучшее и быстрое фильтрование и частичную промывку. Следует не отводить палочку от воронки или от стакана с осадком и не класть ее на стол, чтобы не потерять ни капли раствора. При фильтровании важно не потерять осадок и не добавить вес за счет фильтров. Поэтому используют беззольные фильтры, а не обычную фильтровальную бумагу.
Т. е. фильтры предварительно подготовленные из которых удалена значительная часть минеральных в-в. Масса золы такого фильтра настолько мала, что ею обычно пренебрегают. Обычно масса золы указывается на обложке упаковки фильтров, если она составляет более 0,0002 г, то есть находится в пределах чувствительности аналитических весов, ее отнимают от массы прокаленного осадка. Перед началом фильтрации выбирают фильтр нужной плотности и наиболее подходящего размера. Черная или красная лента - наиболее крупнопористые фильтры и соответственно быстро фильтрующие, используются для фильтрации и отделения аморфных осадков, например, гидроксидов железа, алюминия и др. Белая лента - фильтры средней плотности, применяются для отделения кристаллических осадков. Синяя лента - фильтры мелкопористые используются для отделения мелкокристаллических осадков, сульфата бария, оксалата кальция.
Для последующей промывки учитывают не объем фильтруемой жидкости, а массу определяемого осадка. Он должен заполнять не более половины фильтра, чтобы не возникали проблемы с его промывкой. Используются стеклянные фильтрующие тигли с вакуумной системой, для фильтрования кристаллических осадков. Промывка осуществляется порциями промывной жидкости при ее приливании после профильтровывания предыдущей порции, в воду следует добавлять небольшое кол-во осадителя, чтобы избежать потерь. В случае пептизации, в промывную воду добавляют электролиты-коагуляторы, обычно легко летучие в-ва, удаляющиеся при последующем прокаливании осадка - обычно это растворы, содержащие аммоний.
Осадок увлекает с собой посторонние в-ва, находящиеся вр-ре, это наз-ся соосаждением и предтавлен 4-мя видами: окклюзией, изоморфное соосаждение, соосаждение сообразованием химических соединений, образющихсяя между посторонним в-вом и осаждаемым в-вом, соосаждение в р-те поверхностной адсорбции примесей осадком.
- 1-ый тип - это процесс захвата микрокропримесей внутрь растущих кристаллов осадка и наиболее трудно устраняется.
- 2-ой - процесс образования «смешанных кристаллов» ионами основного компонента и микропримеси, имеющими близкие радиусы, например, сульфат бария может увлекать кристаллы перманганата калия, т.к. эти в-ва изоморфны, т. е. образуют совместную пространственную кристаллическую решетку.
- 3 - ий вид происходит между осаждаемым в-вом и присутствующими в р-ре примесями, например, при осаждении ионов бария раствором серной к-ты, то, в присутствии ионов железа образуется комплексная соединения сульвата бария и железа, при этом следует предварительно гидроксидом аммония удалить примеси железа в виде гидроксида железа, отфильтровав последний.
Иногда для удаления примесей используют переосаждение, т. е. осадок, содержащий примесит, например, оксалат кальция и примесь оксалат магния, растворяют в соляной к-те, нейтрализуют и вновь осаждают оксалатом аммония, т.к. концентрация магния становится меньше, то он не влияет на осаждение кальция. 4-ый вид устраняется промывкой и горячей водой, т. к. адсорбция сопровождается десорбцией. Последняя промывка сопровождается обтиранием стеклянной палочки кусочком фильтра и он кладется в в общий фильтр. И затем делают пробу на полноту удаления примесей, из фильтрата берут 2-3 мл р-ра и проводят качественную реакцию, например на хлориды и т.д. Фильтрат затем отбрасывают, если он не содержит основного в-ва и совсем прозрачен. Отделение осадка и прокаливание. После промывки на фильтре остается практически чистый осадок и остается только узнать его массу.
Т. к. количественно снять осадок с фильтра невозможно, в большинстве случаев вместо этого фильтр подсушивают в сушильном шкафу на воронке при 90 - 1050 С, переносят в предварительно доведенный до постоянного веса фарфоровый, металлический или платиновый тигль и сжигают с предварительным озолением на электроплитке и в течение 20 -30 мин в муфельной печи (в случае, если осадок не взаимодействует с углеродом) или без фильтра, высыпают осадок на глянцевую бумагу и накрывают химическим стаканом или воронкой. Фильтр с остатками осадка сжигают в тигле (постоянный вес), добавляют осадок и прокаливают до постоянного веса.
Расчет ведут в 2-ва приема:
- Сначала узнают, ск. г определяемой составной части содержится в полученном кол-ве весовой формы. Эту величину находят из соотношения молекулярных или атомных весов определяемой составной части и весовой формы.
- Затем (как и в первых 2-х случаях) узнают, сколько % составляет масса определяемой составной части от массы всей взятой навески. Температура прокаливания может ориентировочно определяться по цвету каления муфельной печи, от темно-красной - 525 до ослепительно-белой 1400-15000 С.
Фактор пересчета или аналитический фактор: отношение молярной массы определяемого в-ва к молярной массе в-ва, находящегося в осадке Ф= М опред-го в-ва / М в-ва, нах-ся в осадке. Фактор пересчета показывает сколько граммов определяемого в-ва содержит1 г осадка. Например Ф = М (Ва) М (Ва О4) = 137,40 /233,40= 0,5887. По готовой формуле можно вычислить содержание элемента в сложном веществе: % = ( Ф/ )100, масса полученного осадка,; Ф - фактор пересчета, - навеска исследуемого вещества.
ТИТРИМЕТРИЧЕСКИЕ (ОБЪЕМНЫЕ) МЕТОДЫ АНАЛИЗА, точное измерение объемов веществТЕМА 8
Титриметрический метод основан на точном измерении объемов веществ, вступающих в химическую реакцию. В этом методе используют растворы реактивов точно известной концентрации - титранты. Процесс медленного прибавления титранта к раствору исследуемого вещества называется титрованием. Момент титрования, когда количество прибавленного титранта становится эквивалентным количеству определяемого вещества, называется эквивалентной точкой титрования или точкой эквивалентности. Её находят с помощью индикаторов или по изменению физико-химических характеристик титруемого раствора. Титриметрический анализ отличается быстротой и точностью полученных результатов.
Техника выполнения титриметрического анализа.
Приготовление раствора по точной навеске вещества. Молярные растворы.
Для приготовления 1 л 1 М раствора вещества подсчитывают его молекулярную массу и вычисляют массу навески по формуле: g = ММ1V/1000, где g - навеска вещества, г; М - молекулярная масса, г; М1 - требуемая молярность раствора; V - вместимость мерной колбы, мл. Нормальные растворы. Навеску вычисляют по формуле g = ЭNV / 1000, где Э - эквивалентная масса вещества, г; N -требуемая нормальность раствора; V - вместимость мерной колбы, мл. Установка титра раствора.
Для установки титра растворов, приготовленных по точной навеске, применяют 2-ва способа: отдельных навесок и пипетирования. Более точный, но требующий большего времени для выполнения - способ отдельных навесок. Для его выполнения вычисляют навеску установочного вещества по формуле (см. выше). При этом объем V обычно составляет 25 мл.
Затем на аналитических весах взвешивают три навески (равные вычисленной массе) с точностью до 0,0001 г. Эти навески переносят в чистые колбы и растворяют в дистиллированной воде (по 0,25 мл) или в отдельных случаях в кислоте. После полного растворения навески растворы титруют раствором титранта, титр которого необходимо установить.
Расчет титра раствора титранта (Тт) проводят по формуле
Т = Этgу.в./ Эу..в..Vт.,
- где Эт - эквивалентная масса титранта,
- Эу.в.-эквивалентная масса установочного вещества,
- gу.в.-навеска установочного вещества,
- Vт объем титранта, затраченного на титрование навески, мл.
В 3-х опытах титр рассчитывают для каждой навески отдельно и по найденным значениям определяют его среднее значение и по полученной величине находят среднее значение нормальности Nпр и коэффициента нормальности К титранта по формулам
пр= Тт1000 /Эт; К =Nпр / Nтеор.,
где Nтеор - расчетная нормальность титранта. В способе пипетирования предварительно рассчитывают навеску установочного вещества по формуле g = ЭNV / 1000, приняв V = 250 мл. Навеску установочного вещества взвешивают на аналитических весах с точностью до 0,0001 г, переводят в мерную колбу вместимостью 250 мл, растворяют. Доводят водой до метки водой и тщательно перемешивают. Помещают в колбу для титрования 25 мл раствора, добавляют индикатор и титруют раствором титранта до изменения окраски индикатора.
Нормальность рассчитывают по формуле
пр= Nу.в.25 / Vт,
где Nу.в.- нормальность раствора установочного вещества; Vт - объем титранта, мл. Перечень веществ, применяемых для установки титра важнейших титрантов, и их характеристики приводятся в методических инструкциях и в табл.
113 Справочного руководства по химии, А. И. Артеменко. - М., Высшая школа, 2002 г. Установочные вещества необходимо брать в виде препаратов марки «ХЧ» после их перекристаллизации из воды.
При установке титра способом пипетирования целесообразно использовать в качестве установочного вещества фиксанал соответствующего реагента. Метод нейтрализации применяется для определения содержания различных кислот, оснований, кислых и гидролизующихся солей. В основе этого метода лежит реакция Н+ + ОН- = Н2О.
Кривые титрования метода нейтрализации. В процессе титрования растворов кислот и оснований рН титруемого раствора непрерывно меняется. Сущность метода. В водных растворах это реакция нейтрализации. Н3О++ОН-=2Н2О. Титрантами метода являются растворы сильных кислот и оснований: НС1, Н2ЅО4, NаОН, КОН. Эти вещества не соответствуют требованиям, предъявляемым к стандартным веществам, поэтому концентрацию титрантов устанавливают стандартизацией их растворов. В качестве первичных стандартов чаще всего используют буру Nа2В4О7.10Н2О, безводный карбонат натрия Nа2СО3, дигидрат щавелевой кислоты Н2С2О4.2Н2О. В процессе титрования растворов кислот или оснований рН титруемого раствора непрерывно меняется.
График зависимости рН раствора от количества прибавленного титранта наз-ся кривой титрования. Кривые титрования дают возможность проследить изменение рН раствора в различные моменты титрования, изучить влияние температуры и концентрации реагирующих веществ на процесс нейтрализации, установить конец титрования и помогают сделать правильный выбор индикатора.
Построение кривой титрования сводится к вычислению рН и концентрации титруемого вещества, соли и избыточного титранта в точках, когда к 100 мл титруемого раствора прибавлено 0; 10; 50; 90; 99; 100; 101; 110; 150; 190; 200 мл титранта.
Полученные данные наносят на график, на оси абсцисс которого отложен объем прибавленного титранта, а на оси ординат - соответствующее ему значение рН, причем рН 14 должно находиться в начале координат, а рН 0 - вверху оси ординат. Полученные точки соединяют плавной кривой по лекалу. Рекомендуемый масштаб изображения: по оси х - ось абсцисс -1см соответствует 10 мл, а по оси у - ось ординат - одной 1 рН.
Вычисления концентрации компонентов производят по формулам:
- в точках 5-99 мл с = (100-V/100+V)N;
- в точке 100 мл с = (V/100 +V)N;
- в точках 101 -200 мл с = (V-100/100+V)N,
где V - объем прибавленного титранта, мл; N -нормальность раствора.
В нулевой точке концентрация титруемого раствора задается условиями задачи. рН рассчитывается по формулам: для сильных кислот:
рН = рсα (1),
т.е. рН равняется отрицательному десятичному логарифму концентрации к-ты; для сильных гидроксидов
КОН, NаОН, Са(ОН)2 - рН =14-рсα (2);
для слабых кислот -
рН =1/2(рКα +рсα) (3);
для слабого гидроксида -
рН =14-1/2(рКα +рсα) (4);
смесь растворов слабой кислоты и ее соли (кислотный буферный р-р) -
СН3СООН и СН3СООNа - рН =рКα +рсα - рсb (5);
смесь растворов слабого гидроксида и его соли (щелочной буферный
р-р) - NН4ОН, NН4С1 - рН =14-рКb - рсb +рсс (6);
для солей, образованных слабым гидроксидом и сильной к-ой -
СuЅО4, NН4С1 - рН = 7 -1/2 рКb + 1/2рсс (7);
для солей, образованных слабой кислотой и сильным гидроксидом -
а2СО3, КF - рН = 7 -1/2рКα-1/2рсс (8);
для солей, образованных слабой к-ой и слабым гидроксидом - рН
=7+1/2рКα+1/2рКb (9),
где Кα и Кb - константы электролитической диссоциации кислоты и основания; рКα и рКb - отрицательные логарифмы констант электролитической диссоциации кислот и гидроксидов; сс - концентрация соли, рсс - отрицательный логарифм концентрации соли. Первые два показателя находят по табличным данным и подставляют в соответствующие формулы. Пример.
Вычислить рН 0,1 М р-ра уксусной к-ты. Уксусная к-та - слабый электролит и расчет производим по формуле
рН = ½(рКα+рсα), рКα
для уксусной к-ты равно 4,75. Концентрацию к-ты 0,1 М записываем в виде 10-1, тогда рсс = -1g 10-1 = 1. Подставив найденные значения в формулу, получим рН = ½(4,75 + 1) = 2,85.
Элементы титриметрического анализа. Основные понятия термины и формулы.
Если известны нормальности 2-х растворов и их объемы, при которых достигается точка эквивалентности, то можно вычислить количества эквивалента кислоты и щелочи:
(1/2Н2SО4)=С(1/2Н2SО4).V(Н2SО4)/1000, n(NаОН) = С (NаОН). V (NаОН) /1000,
где V (Н2SО4) и V(NаОН) выражены в миллилитрах.
Таким образом, при подстановке правых частей 2-х последних уравнений в предыдущее равенство получим очень важное для титриметрического анализа выражение принципа эквивалентности:
С(1/2Н2SО4).V(Н2SО4) =С(NаОН).V(NаОН).
В общем виде для любых случаев титрования это соотношение имеет следующее выражение:
Сн(Х).V(Х)=Сн(Т).V(Т),
где Сн(Х), Сн(Т) - нормальности раствора анализируемого вещества Х и титранта Т, V(Х) и V(Т) - объемы анализируемого вещества и титранта соответственно.
Из этого соотношения следует:
(Т)/V(Х)= Сн(Х)/Сн(Т).
Если известен объем анализируемого раствора, то по последнему уравнению можно рассчитать его нормальность:
Сн(Х)= Сн(Т).V(Т)/V(Т).
Вычислив по результатам титрования нормальность анализируемого раствора из вышеприведенных уравнений можно определить массу вещества в любом объеме раствора.
При рассмотрении большинства химических реакций удобно пользоваться понятиями эквивалента и фактора эквивалентности.
Эквивалент - это некая реальная или условная частица, которая может присоединять или высвобождать один ион водорода в кислотно-основных реакциях или один электрон в окислительно- восстановительных реакциях.
В другой формулировке эквивалент - это некая реальная или условная частица, которая в реакции обмена или окисления-восстановления взаимодействует с носителем одного элементарного заряда. Например, в реакции Н3РО4+ ОН- =Н2РО4- +Н2О молекула Н3РО4 равна эквиваленту, т. к. она реагирует с одним ионом ОН-. А в реакции Н3РО4+2ОН- =НРО42-+2Н2О молекула Н3РО4 реагирует с 2-мя ионами ОН-, значит, здесь эквивалент фосфорной кислоты равен ½ молекулы Н3РО4.
Фактор эквивалентности ƒэкв(Х) - это число, показывающее, какая доля реальной частицы вещества Х эквивалента равна одному иону водорода в данной кислотно-основной реакции или одному электрону в окислительно-восстановительной реакции. Это величина безразмерная, рассчитывается на основании стехиометрических коэффициентов конкретной реакции.
Фактор эквивалентности часто обозначают отношением 1/z, где z - суммарный заряд обменивающихся в молекуле ионов для обменных реакций или число электронов, принятых или отданных молекулой (атомом) вещества, для ОВР; z - всегда целое положительное число, а фактор эквивалентности - меньше или равен 1: ƒэкв(Х) = 1/z<1. Фактор эквивалентности одного и того же вещества может иметь разные значения в разных реакциях. В связи с этим, при использовании терминов «эквивалент» и «фактор эквивалентности» вещества всегда необходимо указывать, к какой конкретной реакции они относятся.
Единицей количества вещества эквивалента является моль. Наряду с молярной массой вещества М(Х) в количественном анализе широко пользуются понятием молярной массы эквивалента вещества Х, которой называется масса одного моля эквивалентов этого вещества. Она равна произведению фактора эквивалентности вещества в реакции на молярную массу вещества Х:
Э(Х)=М(ƒэкв(Х)Х)=ƒэкв(Х).М(Х).
Количество вещества (в молях), в котором частицами являются эквиваленты, называется количеством вещества эквивалента(nэ(Х)). Очевидно, что nэ(Х) = m(Х)/Э((Х). Например, требуется рассчитать количество вещества эквивалента безводной соды в ее навеске массой 5,3 г при проведении реакции с НСl до СО2, в соответствии с уравнением реакции Nа2СО3+2НСl =2NаСl +Н2О +СО2, ƒэкв(Nа2СО3)=1/2. В результате уравнения реакции имеем n(1/2Nа2СО3) = m(Nа2СО3)/M(1/2Nа2СО3) =5,3/53 =0,1моль.
Т. о. на основании 2-х последних уравнений легко получить выражение для расчета массы вещества, исходя из количества вещества эквивалента и фактора эквивалентности вещества: M(Х) =nэ(Х).Э(Х) =nэ(Х).ƒэкв(Х).M(Х).
В титриметрическом анализе для выражения состава раствора используют молярную концентрацию эквивалента или нормальность, которая равна количеству вещества эквивалента (в молях), содержащегося в одном литре раствора. Она обозначается С(ƒэкв(Х)Х) (иногда Сн или N с указанием значения фактора эквивалентности вещества) и рассчитывается как отношение количества эквивалента растворенного вещества Х к объему раствора в литрах: Сн(Х)=С(ƒэкв(Х)Х) =nэ(Х)/V = m(Х)/Э(Х).V).
Выразив в знаменателе молярную массу эквивалента через молярную массу вещества и фактор эквивалентности его в реакции по уравнению (1), получим Сн(Х) = m(Х)/(ƒэкв(Х).M(Х).V).
Молярная концентрация равна количеству вещества (в молях), содержащегося в 1л раствора. Она обозначается С(Х), иногда (СM) и рассчитывается как отношение количества растворенного вещества Х к объему V раствора в литрах: С(Х)=n(Х)/V=m(Х)/M(Х).V. При записи молярной концентрации используют, например. следующие формы: раствор с молярной концентрацией НСl, равной 0,1моль/л или С(НСl) =0,1 моль/л, 0,1 М раствор НСl (децимолярный раствор НСl) или 0,1 М НСl (здесь буквой М обозначают «молярный»). Все эти записи означают, что в 1л раствора содержится 0,1моль НСl.
При записи молярной концентрации эквивалента, например, для КMnО4 в полуреакции MnО4-+8Н++5е-=Mn2+ +4 Н2О, используют такие формы: С(1/5 КMnО4) = 0,1моль/л, 0,1 н. или 0,1N раствор КMnО4. Если численные значения молярной концентрации и нормальности совпадают (это наблюдается в тех случаях, когда ƒэкв(Х)=1), то употребляют слово «молярный». Например, для 1М раствора КОН не следует применять выражение 1 н. КОН, а нужно использовать выражение 1 М КОН. Количественная связь между молярной концентрацией вещества и его молярной концентрацией эквивалента выражается уравнением С(Х)/Сн(Х)=ƒэкв(Х). Количество вещества (Х), а следовательно, и его масса в объеме V (л) раствора могут быть рассчитаны как на основе молярной концентрации раствора, так и на основе его нормальности.
В тех случаях, когда речь идет об отношении массы (или объема, или количества вещества) всей системы, термин «концентрация» не употребляют, а говорят о «доле» - массовой, объемной или молярной, и выражают эту долю либо дробью, либо в процентах, принимая систему за 1 или за 100%. Все виды долей в отличие от видов концентрации представляют собой безразмерные величины.
Для обозначения доли компонента приняты следующие греческие буквы: массовая доля ω (омега), объемная доля φ (фи), мольная доля χ (хи), причем: ω((Х)=m(Х)/m, φ(Х)=V(Х)/V, χ(Х) =n(Х)/Σn, где m(Х), m - массы компонента и всей системы соответственно; V(Х), V - объемы компонента и всей системы; n(Х), Σn - количество вещества компонента и всей системы. Следует говорить: «Раствор с массовой долей растворенного вещества 20%». Моляльность раствора - количество вещества (в молях), растворенное в 1 кг растворителя, обозначается Сm(Х) и рассчитывается как отношение количества вещества (Х) к массе растворителя в килограммах: Сm(Х)= n (Х)/m(Υ)=m(Х)/M(Х).mΥ. Титр - ранее использовавшийся способ выражения состава раствора - показывает число граммов растворенного вещества в 1мл раствора.
Раствор точно известной концентрации называется стандартным раствором или титрантом, а процесс постепенного добавления его к анализируемой пробе - титрованием. Момент завершения реакции между титрантом и определяемым веществом называется точкой стехиометричности (точкой эквивалентности).
К реакциям, протекающим в стехиометрических отношениях, применим закон эквивалентов. Поэтому, если реакция проведена до конца, число эквивалентов определяемого компонента равно числу эквивалентов реагента. Т. е. моль эквивалента любой кислоты способен нейтрализовать моль эквивалента любого основания. Таким образом, и при титровании, поскольку его заканчивают в точке эквивалентности, всегда затрачиваются одинаковые количества эквивалента титруемого и титрующего веществ. Например, в точке эквивалентности титрования раствора Н2SО4 раствором щелочи (Н2SО4 +2NаОН =Nа2SО4 +2Н2О) количества эквивалентов Н2SО4 и NаОН равны между собой: n(1/2Н2SО4=n(NаОН).
Методы титриметрического анализа.
В точке эквивалентности количество вещества щелочи, израсходованное на реакцию, всегда точно равно количеству вещества кислоты в анализируемом растворе, например: n(NаОН)=n(НСl) или в общем виде, для любых реагирующих веществ по закону эквивалентов: n1=n2. Для понимания сущности титриметрического анализа и связанных с ним расчетов важное значение имеет уравнение: с(NаОН)V(NаОН)=с(НС1)V(НС1), где с -нормальные концентрации растворов; V - их объемы. Общее уравнение имеет вид с1V1=с2 V2. Если известен объем одного из растворов, то из этого уравнения можно вычислить его концентрацию: с1=с2V2/V1 или с2=с1V1/ V2.Точку эквивалентности обычно устанавливают по изменению окраски индикатора (индикаторный способ), но иногда прибегают к измерению электрической проводимости или других свойств раствора (физико-химические) способы. Достигнув точки эквивалентности, титрование прекращают. По затраченному объему титранта и его концентрации вычисляют результаты анализа. Предположим, необходимо определить содержание гидроксида натрия в растворе. Для этого отмеряем в коническую колбу точный объем анализируемого раствора (например, 10,00 мл) и титруем из бюретки титрантом - титрованным раствором НС1. В примере титр кислоты равен 0,002302г/мл и до достижения точки эквивалентности добавили 17,50 мл раствора кислоты.
Следовательно, на реакцию израсходовали
17,50.0)002302=0,04029 г НС1.
По уравнению реакции нетрудно вычислить какой массе NаОН, находящегося в растворе, соответствует эта масса кислоты:
40 г - 36,5, а 0,04029 - х, получится 0,03676 г.
Преимущество титриметрического анализа перед гравиметрическим состоит в скорости, т. е. быстроте определения. В гравиметрическом анализе осаждение (выполнение реакции) для получения осаждаемой формы является лишь началом определения, в титриметрическом анализе выполнение реакции (титрование) и заканчивает определение. Практически одинакова и точность этих анализов.
В титриметрическом анализе применяются химические реакции различных типов и в различных практических областях в агрохимии, в пищевых лабораториях, в фармацевтических и различных производственных лабораториях. По типу используемых химических реакций методы титриметрического анализа разделяют на три группы: 1. методы, основанные реакциях соединения ионов; 2. реакциях окисления-восстановления; 3 на реакциях комплексообразования. К 1-ой группе относят методы кислотно-основного и осадительного титрования, ко второй - различные методы окислительно-восстановительного титрования и к 3-ей - методы комплексометрического (хелатометрического) титрования.
Метод кислотно-основного титрования (нейтрализации) основан на взаимодействии кислот с основаниями или на взаимодействии протонов с гидроксидионами: Н+ + ОН- = Н2О +57,32Дж. Метод позволяет определять в растворах не только концентрацию кислот и оснований, но и концентрацию гидролизующихся солей. Для определения в растворах концентрации оснований или солей, дающих при протолизе щелочную реакцию, используют титрованные растворы кислот - ацидиметрию. Концентрацию кислот или гидролитически кислых солей определяют с помощью титрованных растворов сильных оснований - алкалиметрия. Точку эквивалентности при нейтрализации определяют по изменению окраски индикатора. Метод осадительного титрования - определяемый элемент при взаимодействии с титрованным раствором, может осаждаться в виде малорастворимого соединения. Последнее, изменяя свойства среды, позволяет тем или иным способом определить точку эквивалентности - аргентометрия с индикатором хроматом калия для определения ионов хлора, до выпадения кирпичнокрасного осадка хромата серебра. В этот момент титрование прекращают и ведут расчет. Название метода определяется раствором титранта: аргентометрия, тиоцианатометрия NН4SСN.
Стандартные и стандартизированные растворы.
Титрованные растворы могут быть получены разными способами и поэтому различают стандартные (приготовленные) и стандартизированные (установленные) р-ры. Стандартные р-ры. Точную навеску в-ва переносят в мерную колбу определенной емкости, растворяют и доводят объем р-ра водой до метки. В этом случае титр р-ра равен навеске (m, г), деленной на объем р-ра (V, мл): Т = m/V. Чтобы перейти к молярной концентрации эквивалента (с), достаточно титр рас-ра умножить на 1000 и разделить на молярную массу эквивалента растворенного в-ва. Например, если 0,5312 г карбоната натрия Nа2СО3 растворили в мерной колбе вместимостью 100 мл, то титр раствора равен Т=0,5312/100=0,005312 г/мл, а молярная концентрация эквивалента с=0,005312· 1000/53,0=0,1002. Титрованные растворы, полученные по точной навеске вещества, называют стандартными (приготовленными).
Подобным способом могут быть приготовлены р-ры, отвечающие определенным требованиям: они должны быть хч; устойчивыми при хранении как в твердом виде, так и в растворе; состав их должен строго соответствовать определенной химической формуле. Такие р-ры называют первичными стандартами для установления титра других рабочих растворов. Примеры, для установки титра кислот служат тетраборат натрия Nа2В4О7· 10Н2О и карбонат натрия Nа2СО3. Титры (или нормальные концентрации) растворов оснований устанавливают по таким исходным стандартным веществам, как щавелевая Н2С2О42Н2О или янтарная Н2С4Н4О4. Таким образом, теперь следует объяснить, что такое стандартизированные растворы. Как можнопонять из вышесказанного к ним относятся вещества, не обладающие вышеперечисленными свойствами стандартных растворов. Это минеральные кислоты, щелочи, перманганат калия, тиосульфат натрия. Точные их растворы не возможно приготовить по навеске, т. к. продажные минеральные к-ты имеют непостоянный состав, а выделить их в чистом виде невозможно. А щелочи NаОН и КОН даже при взвешивании поглощают С (1V) и воду из воздуха, т. е. изменяют свой состав Перманганат калия и тиосульфат изменяются при растворении, взаимодействуя с примесями воды.
Следовательно, содержание их в растворе не может соответствовать навеске. Поэтому аналитики готовят растворы этих веществ, взвешивая приблизительно необходимое количество реагента и растворяя навеску в необходимом объеме воды. Затем титруют подходящим раствором стандартного вещества. Зная объем, пошедший на титрование, и концентрацию стандартного вещества - известную концентрацию раствора первичного стандарта, вычисляют нормальную концентрацию и титр стандартизируемого. Растворы, титр которых находят не по точной навеске, а устанавливают по стандартному веществу, называют установленными или стандартизированными (растворами вторичных стандартов). Для соляной к-ты по раствору тетрабората натрия, для гидроксида натрия по стандартному раствору щавелевой к-ты. Применяют идругие приемы, например, устанавливают титр р-ра НС1 путем титрования раствором натрий гидроксида, если нормальная его концентрация установлена предварительно по соответствующему стандартному веществу. Раствор соляной кислоты после такого определения титра может быть также использован для дальнейшего установления титра других веществ. Этот прием очень удобен в практике, т.к. не надо каждый раз готовить стандартные разные растворы, но недостаточно точен и вносит иногда значительную погрешность в анализ. Следовательно, титрованные стандартные растворы следует готовить очень тщательно и пользоваться одной и той же мерной посудой как при их приготовлении, так и при проведении анализа, т. е. в одинаковых условиях. В практике титрованные растворы готовят из фиксаналов или стандарт-титров. Это запаянная стеклянная ампула содержащая количество вещества необходимое для приготовления 1 л точно 0,1 н или 0,01н раствора. Фиксаналы выпускаются в виде жидких (минеральные к-ты, едкие щелочи) и сухих (перманганат калия, карбонат или оксалат натрия и др.).
Рассказать, как готовят раствор из фиксанала (боек, воронка, промывалка). Титрованные растворы обычно хранятся несколько месяцев, если нет специальных указаний, особенно требовательны в этом отношении едкие щелочи, при хранении их необходимо предохранять от контакта с воздухом, их хранят в полиэтиленовой плотно закрытой посуде. Для титрования стандартными растворами есть специальные столы и титровальные установки, которые используются при серийных анализах. При этом стандартные или установочные растворы не контактируют с воздухом и с помощью специальных сифонов раствор поступает в бюретку, есть специальные автоматические бюретки. При пользовании мерной посудой существуют определенные правила пользования и мойки. Для бюреток: каждое титрование начинают с конечного деления шкалы, т. к. при этом лучше всего компенсируются погрешности калибрования бюретки.
Выпускают раствор из бюретки не очень быстро (не более 3-4 капель в секунду). Иначе раствор не будет вовремя стекать со стенок и отсчет окажется неверным. Объем расходуемого на тирование р-ра не должен превышать вместимость одной бюретки и не превышать 20 мл для 25 мл бюретки и 30 мл для бюретки на 50 мл. Ошибка на +- 0,02 мл составит 0,02·100 / 10мл = 0,2%. После окончания работы сливают раствор из бюретки, моют, наполняют дистиллированной водой и накрывают пробиркой для предохранения от пыли. Правила заполнения раствором бюретки и удаление пузырьков воздуха. Пипетки. Не следует выдувать или вытряхивать последние капли жидкости из пипетки, моют пипетки обычным способом, перед употреблением ополаскивают рабочим раствором, с которым будут работать. Проверка мерной посуды взвешиванием и бюреток по 5 мл р-ра и с учетом плотности воды и поправки на температуру. Вычисления в титриметрическом анализе.
Главный принцип: вещества реагируют друг с другом всегда в эквивалентных количествах. Так на титрование до точки эквивалентности всегда расходуется одинаковое число эквивалентных масс кислоты и основания. Следовательно при одинаковой нормальной концентрации растворов реагирующих веществ реакции идут между их равными объемами. Например, на титрование 10 мл 0,1н. р-ра всякой кислоты расходуется такой же объем 0,1 н. р-ра любой щелочи. В этом также удобство использования нормальных растворов.
Результаты титриметрического анализа вычисляют тремя способами: 1. Вычисление при выражении концентрации раствора через титр по определяемому веществу; 2. При титровании по методу пипетирования; 3. При титровании методом отдельных навесок. По первому методу, например, Т(АgNО3/Сl-) показывает, сколько граммов С1- осаждается 1 мл р-ра нитрата серебра.
В примере концентрация р-ра нитрата серебра равна 0,1100. Тогда 1 мл его содержит 0,1100: 1000 молярных масс эквивалентов нитрата серебра и взаимодействует с таким же числом молярных масс эквивалента иона хлора. Молярная масса эквивалента хлора равна 35,46 г/моль, вычисляем титр нитрата серебра по хлору: Т(АgNО3/Сl-) = 0,1100. 35,46/1000=0,003901 г/моль. На титрование израсходовали 15,00 мл р-ра нитрата серебра, то титруемый р-р содержит хлора: m (С1-)=Т(АgNО3/Сl-)V(АgNО3) = 0,003901·15,00 = 0,05851 г. При вычислениях по нормальности растворов ход вычислений зависит от метода титрования. Метод пипетирования состоит в том, что навеску анализируемого вещества растворяют в мерной колбе, доводят объем водой до метки и для титрования берут определенные (аликвотные) порции р-ра пипеткой.
Для расчета используют уравнение с1V1=с2V2. Задача: в мерную колбу внесли 0,6504 г продажной щавелевой к-ты. Растворили и довели объем до метки 100 мл. Пипеткой брали по 10 мл полученного р-ра и титровали 0,1026 н. р-ром гидроксида натрия, расход которого составил в среднем 9,85 мл. Определить массовую долю (%) щавелевой к-ты в продажном препарате. Вычисляем нормальную концентрацию раствора щавелевой к-ты: с(Н2С2О42Н2О) 10,00= 0,1026. 9,85; с =0,1026. 9,85/10,00 =0,1011. Затем находим массу щавелевой к-ты в 0,1 л р-ра. При этом исходить следует из того, что щавелевая к-та превращается в щавелевокислый натрий, следовательно, молярная масса эквивалента ее равна ½ молярной массы, т.е.126,6: 2=63,03 г/моль.
Поэтому m=0,1011. 63,03.0,1=0,6372 г. Следовательно, массовая доля (%) щавелевой к-ты равна: ω (Н2С2О42Н2О)= m (Н2С2О42Н2О)/ m(навески)= 0,6372/0,6504=0,9797, или 97,97%. В случае использования метода отдельных навесок уравнение с1V1=с2V2 не может быть использовано, т. к. навеску растворяют в произвольном объеме воды. Вычисляют результат, исходя из того, что при титровании в-ва взаимодействуют эквивалентными количествами. Какова массовая доля (%) Н2С2О42Н2О в образце щавелевой к-ты, если на титрование 0,1500 г его пошло 25,60 мл 0,09002 н. р-ра NаОН? Вычисляют, сколько молярных масс эквивалента NаОН содержалось в 25,60 мл раствора и участвовало в реакции: 1000 мл р-ра NаОН содержит 0,09002 молярных масс эквивалента NаОН, а 25,60 мл NаОН содержат х молярных масс NаОН, Х = 25,60. 0,09002 / 1000 = 0,002305 молярных масс эквивалента NаОН. Но при титровании 1 эквивалент в-ва взаимодействует с 1 эквивалентом другого. Т. е. Это означает, что в реакции участвовало также 0,002305 молярных масс эквивалента Н2С2О42Н2О. Молярная масса эквивалента щавелевой к-ты равна 63,03 г/моль. Следовательно, анализируемый раствор содержит 0,002305. 63,03 = 0,1453 г Н2С2О42Н2О. В массовых долях (%) это составляет: ω (Н2С2О42Н2О) = m (Н2С2О42Н2О)/m (образца) = 1,1435 г / 0,1500 г = 0,9686 (96,86%). Во многих случаях для уточнения результатов анализа (из-за неточности навесок при приготовлении растворов или исследуемого вещества) вводят понятие поправочного коэффициента.
Поправочный коэффициент К равен отношению практически взятой навески к теоретически вычисленной К = С/ С1, где С - нормальность приготовленного раствора; С1 - теоретическая нормальность. Например, требуется приготовить р-р с заданной (теоретической) концентрацией 0,1000 моль/л. Практически, приготовлен раствор с близкой концентрацией 0, 1056 моль/л. Тогда находим поправочный коэффициент К = 0,1056 / 0,1000 = 1,056. Поэтому практически во всех титриметрических анализах, в расчетах вводится поправочный коэффициент для данного раствора. При массовых анализах и использовании нового раствора (вновь приготовленного) вводится новый поправочный коэффициент, при правильном приготовлении растворов он должен быть в пределах 0,98 - 1,02.
Следовательно, для расчетов следующие основные правила:
. Титры раствороов, выраженные в граммах различных в-в относятся друг к другу как молярные массы эквивалентов этих в-в: ТН2SО4/ NаОН/ТН2SО4=Э NаОН/ЭН 2 SО 4, ТН 2 SО 4 / NаОН = ТН2SО4.ЭNаОН/ЭН2 SО4, где Т Н2 SО4/NаОН - титр по определяемому в-ву, т. е. количество граммов определяемого в-ва, оттитрованное 1 мл определяемого раствора. При одинаковой концентрации р-ов реакции идут между равными объемами их: V1/ V2=С2/С1 или V1/ V2= N2/ N1.
Массу навески в-ва можно вычислить по формуле:
q = СVЭ / 1000.
МЕТОДЫ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ, реакции нейтрализации, скачок титрования, построение кривых титрованияТЕМА 9
Кислотно-основное титрование, реакции нейтрализации, скачок титрования, построение кривых титрования, правила выбора индикатора при кислотно - основном титровании, требования, предъявляемые к индикаторам, методы амперометрического, кондуктометрического и потенциометрического титрования.
Кривая титрования
графическое изображение зависимости изменения концентрации С (Х), определяемого в-ва Х или некоторого связанного с ним свойства системы (раствора) от объема V(Т) прибавленного титранта (Т).
По оси абсцисс откладывают объем прибавляемого титранта, по оси ординат соответствующее значение рН. Потенциометрическое тирование - изменение концентрации иона непременно сопровождается изменением потенциала на индикаторном электроде, погруженном в титруемый р-р.
При этом около точки эквивалентности наблюдается скачок потенциала, который фиксируется при помощи потенциометра. Кондуктометрическое титрование - изменение электрической проводимости титруемой среды между 2-мя инертными электродами. Амперометрическое титрование - за ходом титрования следят с помощью ртутного капающего, вращающегося платинового или другого микроэлектрода, играющего роль индикаторного электрода и находящегося в паре с подходящим электродом сравнения.
Кулонометрическое титрование-измерение количества электричества, затраченного на выполнение электродной реакции. При этом титрующий агент (например. кислотно-основного типа) не прибавляется в виде стандартного р-ра, а образуется в р-те электродной реакции, т. е в результате электролиза при постоянном токе.
Титрование сильной кислоты сильным основанием:
НС1+КОН =КС1+Н2О,
в процессе титрования происходит постоянная, непрерывная нейтрализация к-ты. В точке «100» наступает полная нейтрализация. Т. к. соль сильного основания и сильной к-ты не способна к гидролизу, рН в этой точке равен 7 (кривая титрования на обороте).
При дальнейшем титровании в р-ре накапливается избыток щелочи и в этом случае рН р-ра рассчитывается по формулам 1 и 2. Титрование слабой к-ты сильным основанием:
СН3СООН+КОН=СН3СООК+Н2О,
в нулевой точке рН вычисляют по формуле 3. По мере титрования в среде накапливается ацетат натрия. Смесь слабой к-ты и ее соли представляет собой кислотный буферный р-р. Поэтому в интервале 10-99 мл прибавленного титранта рН рассчитывают по формуле 5. В точке 100 вся к-та полностью связана в соль. Соль слабой к-ты и сильного основания легко гидролизуется с отщеплением свободных гидроксилов.
Поэтому точка эквивалентности находится в щелочной области (кривая титрования на обороте); рН рассчитывается по формуле 8. При дальнейшем титровании в растворе накапливается избыток титранта и рН в области 101-200 мл прибавленного титранта рассчитывают по формуле.
Титрование слабого основания сильной кислотой:
NН4ОН+НС1=NН4С1+Н2О,
в нулевой точке рН раствора вычисляют по формуле 4. По мере титрования в р-ре накапливается соль - хлорид аммония. Смесь слабого основания и его соли представляет собой щелочной буферный р-р, поэтому расчет рН в интервале 10-99 мл прибавленного титранта проводят по формуле 6. В точке эквивалентности (точка 100) все основание переходит в соль. Соль слабого основания и сильной к-ты легко гидролизуется с отщеплением свободных протонов. Поэтому точка эквивалентности находится в кислой области (кривая тирования на обороте), расчет рН ведут по формуле 7.
При дальнейшем титровании в р-ре накапливается избыток титранта и рН в интервале 101-200 рассчитывается по формуле 1. Титрование слабой к-ты слабым основанием: СН3СООН+NН4ОН =СН3СООNН4+Н2О. В нулевой точке рН раствора находят по формуле 3. В интервале 10-99 мл прибавленного титранта смесь представляет собой кислотный буфер и рН р-ра рассчитывают по формуле 5. В точке эквивалентности вся кислота связана в соль. Соль слабой к-ты и слабого основания полностью гидролизуется с образованием слабых к-ты и основания. В зависимости от значений рКα и рКb рН р-ра соли может находиться как в кислой, так и в щелочной области (кривая титрования); рН в этой точке рассчитывается по формуле 9.
При дальнейшем тировании рН в точке 101 находят по формуле 4. После того как в растворе накопится достаточный избыток титранта, титруемая смесь будет представлять собой щелочной буфер (смесь слабого основания и его соли) и рН в интервале 110-200 мл вычисляют по формуле 6. Как мы уже говорили ацидиметрия - ацидиметрическое тирование - это метод определения сильных и слабых оснований, солей слабых к-т, основных солей и других соединений, обладающих основными свойствами, путем титрования стандартным р-ром сильной к-ты. При титровании сильных оснований протекает реакция:
ОН-+Н3О+=2Н2О, среда в ТЭ - нейтральная.
При титровании слабых оснований, например аммиака, NН3+Н3О+= NН4++Н2О образуются катионы слабого основания, подвергающиеся гидролизу:
NН4++Н2О= NН3+Н3О+
поэтому среда в ТЭ - слабокислая, рН<7. При титровании солей слабых одноосновных к-т, например ацетатов,
СН3СОО-+Н3О+= СН3СООН+Н2О в ТЭ в р-ре присутствует слабая к-та, вследствие диссоциации которой р-р имеет слабокислую реакцию, рН<7.
При титровании солей слабых 2-хосновных к-т, например карбонатов, после присоединения к аниону к-ты одного протона образуется кислый анион слабой к-ты СО32-+Н3О+=НСО3-+Н2О, подвергающийся гидролизу: НСО3-+Н2О=Н2СО3+ОН-, вследствие чего реакция среды в 1-ой ТЭ -слабощелочная, рН>7. При продолжении титрования кислого аниона слабой 2-хосновной к-ты во 2-ой ТЭ присутствует эта слабая к-та: НСО3-+Н3О+=Н2СО3 вследствие частичной диссоциации которой среда во 2-ой ТЭ - слабокислая, рН<7. Т.о. при ацидиметрическом титровании среда в ТЭ может быть нейтральной, слабо щелочной или слабокислой в зависимости от природы титруемого вещества.
И далее при титровании до 2-ой ТЭ в среде присутствует анион ЅО32- слабой к-ты НЅО3-: НЅО3-+ОН-=Н2О+ЅО32-, этот анион подвергается гидролизу: ЅО32-+Н2О=ОН-+НЅО3- и среда во 2-ой ТЭ - щелочная, рН>7. При титровании кислых солей образуются средние анионы соответствующей кислоты, свойства которых и определяют значение рН среды в ТЭ. Например, при титровании р-ра NаНЅО4 щелочью образуется сульфат-ион ЅО42-: НЅО4-+ОН-=Н2О+ЅО42-. Сульфат ион практически не подвергается гидролизу, поэтому среда в ТЭ - нейтральная. Соли, содержащие катионы слабых оснований, титруются щелочами с выделением свободного основания. В ТЭ присутствует это основание, поэтому и реакция среды в ТЭ - слабощелочная. Индикаторы кислотно-основного титрования. Индикатор - это в-во, которое проявляет видимое изменение в точке эквивалентности или вблизи ее. При визуальном индикаторном методе фиксации КТТ в кислотно-основном титровании прибавление титранта к титруемому раствору прекращают, когда резко меняется окраска раствора вследствие изменения цвета индикатора, введенного в титруемый р-р.
Показатель титрования рТ - значение рН, при котором становится заметным изменение окраски индикатора и заканчивается титрование. Кислотно - основные индикаторы - слабые органические к-ты, диссоциирующие в растворах по уравнению Н1nd ↔Н++1nd-. Окраска молекулярной (Н1n) и анионной (1nd)- форм различаются. Это связано с тем, что в процессе диссоциации индикатора происходит таутомерная перегруппировка, приводящая к изменению строения молекулы, обуславливающего окраску соединения. Существует несколько теорий, объясняющих изменение окраски индикаторов, одна из них хромофорная, т. е. окраска в-в зависит от наличия в их молекуле особых атомных группировок - хромофоров. Хромофор в органических соединениях чаще всего представляет собой систему сопряженных (чередующихся) простых и двойных связей. Чем больше длина цепи сопряжения, тем более сильно окрашено в-во.
В кислотно-основных индикаторах важнейшими хромофорами являются азогруппа -N = N-, хиноидное кольцо и хинониминное кольцо. В процессе таутомерной перегруппировки хромофорная группа либо разрушается, вследствие чего индикатор обесцвечивается, либо превращается в другой хромофор, в результате чего индикатор меняет свой цвет. Первый тип таутомерной перегруппировки характерен для одноцветных индикаторов (типа фенофталеина), второй - для 2-х цветных индикаторов (типа метилового оранжевого). Последний в водных р-ах анион метилового оранжевого имеет желтую окраску, а в присутствии кислот - красную. При этом, происходит разрушение азогруппы и превращение бензольного кольца в хинониминное, что и сопровождается изменением окраски метилового оранжевого. Область рН, в которой происходит изменение окраски, называется интервалом перехода индикатора ΔрН=рКн1пd+-1, где рКН1пd - показатель константы диссоциации индикатора.
Выбор индикатора для титрования проводится по кривой титрования 2-мя способами:
- Для данного титрования наиболее пригоден тот индикатор, для которого интервал перехода ΔрН попадает в область скачка на кривой титрования.
- Для данного титрования наиболее пригоден тот индикатор, у которого показатель титрования рТ находится наиболее близко к рН эквивалентной точки титрования.
Кривая, выражающая изменение значения рН титруемого раствора в зависимости от объема прилитого рабочего раствора, наз-ся кривой титрования. При сравнении кривых титрования, т.е. ветвей кривой титрования, расположенных выше и ниже точки эквивалентности, видно, что при титровании сильных растворов ветви более крутые, чем при титровании слабых р-ров. У слабых растворов - сглаженные ветви. В первом случае ветви кривых соответствующие сильным компонентам увеличивает область скачка, в противном случае - происходит уменьшение скачка.
Поэтому кривая α с обеими ветвями 1-го типа имеет самый большой скачок титрования. Кривые б и в состоят из разных ветвей 1 и 11 типа и поэтому имеют гораздо меньший скачок. Т.о., в случае слабого компонента скачок всегда уменьшается. Если же титровать слабое основание слабой кислотой, то кривая титрования будет состоять из обоих ветвей 11-го типа и совсем не будет иметь вертикальной части, т.е. в этом случае не будет наблюдаться заметный скачок титрования. Поэтому при титровании слабого основания слабой кислотой рН титруемого раствора изменяется настолько постепенно, что установить точно момент, когда следует прекратить титрование (т.е. точку эквивалентности) практически невозможно.
Поэтому такое титрование практически не применяют. Построить кривую титрования - это значит нанести на график следующие точки: 1) исходную точку титрования - соответствующую значению рН исходного титруемого р-ра до начала титрования. Эта точка находится всегда на вертикальной линии, соответствующей 0 мл рабочего р-ра; 2) точку эквивалентности, показывающую, при каком значении рН должно быть закончено титрование. Эта точка всегда лежит на линии эквивалентности; 3) нанести промежуточные точки кривой, показывающие, как изменяется рН р-ра в процессе титрования.
Для кривой титрования сильной к-ты сильным основанием характеризуется следующими особенностями:
- . Исходная точка титрования лежит в сильно кислой среде.
- . Точка эквивалентности лежит на линии нейтральности.
- . В начале титрования рН изменяется очень медленно. Резкое изменение начинается только при приливании последней 0,1 части рабочего р-ра.
. Скачок титрования (при титровании 0,1 н. р-ра кислоты) очень велик - от рН=4 до рН=10. Если титрование идет в обратном порядке, т.е. в колбе находилась бы щелочь, а в бюретке к-та, то кривая титрования сохранила бы тот же вид, но исходная точка титрования находилась бы в нижней части графика при рН=13.
Определение рН при помощи индикатора, стр. 14 -15. приступая к титрованию, необходимо сначала рассчитать значение рН, при котором следует закончить титрование р-ра. В соответствии с этим значением рН и выбирают индикатор, т. к. нужно, чтобы индикатор изменял цвет при значении рН, отвечающем точке эквивалентности данного титрования. В действительности в этом нет необходимости, т. к. кривые титрования показывают, что ТЭ лежит приблизительно в середине скачка рН, а этот скачок наступает при изменении рН р-ра от прибавления последней капли рабочего р-ра. Поэтому от прибавления последней капли изменяют цвет все индикаторы, интервал перехода которых соответствует скачку титрования. Т. о. главное правило выбора индикатора: титрование в методе нейтрализации следует выполнять в присутствии такого индикатора, интервал перехода которого заключается в интервале скачка рН на кривой титрования данного в-ва.
В случае титр-ия слабой к-ты сильным основанием скачок титрования от 8 до 10 ед. рН, что соответствует фенолфталеину, в случае сильной к-ты и сильного основания - скачок от 4 до 10 и выбор индикатор тут достаточно широк. Титрование со «свидетелем» применяется для точности определения цвета титруемого р-ра, т.е. используется образцовый р-р, имеющий такую же окраску, как и та, при которой нужно закончить титрование (одинаковый сосуд, объем и индикатор в кол-ве по 1капле на 25 мл р-ра). Оксидиметрия. Окисдительно-восстановительные методы объемного анализа основаны на применении ОВР.
Рабочими р-ми являются р-ры окислителей или восстановителей. Метод, основанный на окислении перманганатом, наз-ся перманганатометрией, на окислении иодом -иодометрией, на окислении хроматом - хроматометрией. ОВП - это колич-ая хар-ка интенсивности окислительно-восстановительного процесса (в вольтах), реакирующих между собой систем. ОВПотенциал систем, измеренный при условии, что концентрации (точнее активности) ионов восстановленной и окисленной форм равны 1, наз-ся нормальным окислительно-восстановительным потенциалом системы. В любой ОВР участвуют, по крайней мере, 2-ве редокс-пары. Rеd1+Ох2=Ох1+Rеd2. Восстановленная форма одного в-ва Rеd1, отдавая электроны, переходит в окисленную форму Ох1 того же в-ва. Обе эти формы образуют одну редокс пару Ох1|Rеd1. Окисленная форма Ох2 второго в-ва, принимая электроны, переходит в восстановленную форму Rеd2 того же в-ва. Обе эти формы также образуют редокс пару Ох2|Rеd2. Чем выше ОВП редокс пары Ох2|Rеd2, окисленная форма которой играет роль окислителя в данной реакции, тем большее число восстановителей Rеd1 можно оттитровать и определить с помощью данного окислителя Ох2.
Поэтому в редоксметрии в качестве титрантов чаще всего применяют окислители, стандартные ОВ потенциалы редокс пар которых имеют как можно более высокие значения. Известно н-ко 10-ов различных методов ОВ титрования. Обычно их классифицируют следующим образом: по характеру титранта - оксидиметрия, методы определения восстановителей с применением титранта-окислителя; редуктометрия - методы определения окислителей с применением титранта-восстановителя. По природе реагента, взаимодействующего с определяемым в-вом, например, иодатометрия (иодат калия К1О3), иодиметрия (иод 12), иодометрия (иодид калия К1). Как известно из теории кач-го анализа зависимость ОВП от концентрации ионов окисленной и восстановленной форм выражается ур-ем Нернста. В случае перманганатометрии главным рабочим р-ром является перманганат калия. Окисление перманганатом с аналит-ми целями проводят преимущественно в кислой среде.
Поэтому основное ур-ие перманганатометрии - МnО4- +5е +8Н+---->Мn2++4Н2О, т.е. перманганат-ион, имеющий интенсивную фиолетово-малиновую окраску, восстанавливается до катионов марганца (11), которые обладают очень слабой розоватой окраской (практически бесцветны). По ур-ию Нернста: Е=Е0+(0,059/5)lg([МпО4-][Н+]5/[Мп2+]. Нормальный потенциал этой системы, равный +1,52 в значительно выше, чем других систем, поэтому КМпО4 в кислой среде обладает сильными окислительными свойствами и способен окислять очень многие в-ва. Величину грамм-эквивалента окислителя (или восстановителя) находят путем деления его молекулярного веса на число принимаемых (или отдаваемых) им в данной химической реакции электронов. Т. к. их 5, молярная масса эквивалента перманганата равна М/5 =158,04/5 = 31,68 г. В практике используются растворы различной концентрации: 0,01н., 0,05н., 0,2н и в основном 0,1н р-р.
В нейтральной среде перманганат-ион восстанавливается до диоксида марганца МпО2:
МпО4-+3е+2Н2О=МпО2+4ОН-.
В этом случае стандартный ОВП редокс пары МпО4-|МпО2 низок (0,60 в), при титровании выпадает бурый осадок диоксида марганца, что затрудняет фиксацию КТТ. В сильнощелочной среде перманганат-ион восстанавливается до манганат-иона МпО42-: МпО4-+е=МпО42-зеленого цвета с образованием бурого осадка диоксида марганца и перманганат-иона, что также искажает результаты анализа. Следовательно, прманганатометрическое титрование целесообразно проводить в сильнокислых средах. Применение индикатора не требуется и для титрования используют бюретки со стеклянными кранами, т.к. резина взаимодействует с перманганатом калия. Данный вид титрования применяется для определения в-в, играющих по отношению к перманганат-иону роль восстановителей, таких как Н2О2, МgО2, NаNО2, металлическое железо, карбоновые к-ты, для определения общей окисляемости воды и почвы (т.е. для определения суммы восстановителей, присутствующих в этих объектах).
Раствор титранта стандартизируют по установочным растворам, в основном щавелевой к-ты, Н2С2О42Н2О и щавелевокислого натрия. Комплексиметрия (комплексометрия) - метод титриметрического анализа, основанный на использовании реакций комплексообразования между определяемым компонентом анализируемого р-ра и титрантом. Или можно сказать, что данный способ титрования - это титрование в-ва раствором такого соединения, которое образует с титруемым в-вом слабо диссоциирующий (т.е. устойчивый) растворимый комплекс. Метод чаще всего применяется для определения катионов металлов-комплексооброзователей.
Классификация данных методов основана на на природе реагента или образующихся комплексов: меркуриметрия - титрование, основанное на использовании реакций образования растворимых, устойчивых, слабо диссоциирующих комплексов ртути (11); цианометрия - использование реакций образования растворимых, устойчивых, слабо диссоцииирующих цианидных комплексов металлов, содержащих в качестве лигандов цианогруппы серебра, цинка, ртути, кобальта, никеля; комплексонометрия - метод, основанный на использовании реакций образования комплексонатов - комплексных соединений катионов металлов с комплексонами, наиболее распространенный метод. При этом способе титрования в результате реакции между катионом металла и комплексоном образуется комплексонат металла. Это чаще всего многоосновные аминополикарбоновые к-ты и их соли, анионы которых способны образовывать со многими катионами металлов устойчивые растворимые комплексы - комплексонаты. Комплексон 111 или ЭДТА (этилендиаминтетраацетат) или трилон Б.
В качестве титрантов применяют стандартные водные р-ры ЭДТА с конц-ей 0,1; 0,05; 0,025 и 0,01 моль/л, а также стандартные р-ры сульфата магния и свинца. Для визуальной фиксации КТТ применяют индикаторы, т.н. металлохромные индикаторы - органические красители, имеющие свои хромофорные группы, способные обратимо изменять окраску при образовании комплексов с катионами металлов. Комплексонометрически контролируют жесткость воды. Общую жесткость воды (т. е. суммарное содержание в ней катионов магния и кальция, в мг-экв/л) определяют прямым титрованием стандартным р-ром ЭДТА в присутствии индикатора кислотного хромового темно-синего. Метод позволяет раздельно определять катионы металлов при их совместном присутствии, как в методе определения жесткости воды. Т. о. в аналитической химии широко применяются титриметрические методы определения различных в-в и элементов, причем в основе этих методов лежат правила качественного анализа и общие химические законы и свойства химических элементов и соединений.
Если известны нормальности 2-х растворов и их объемы, при которых достигается точка эквивалентности, то можно вычислить количества эквивалента кислоты и щелочи:
- (1/2Н2SО4)=С(1/2Н2SО4).V(Н2SО4)/1000, n(NаОН) = С (NаОН). V (NаОН) /1000,
где V (Н2SО4) и V(NаОН) выражены в миллилитрах.
Таким образом, при подстановке правых частей 2-х последних уравнений в предыдущее равенство получим очень важное для титриметрического анализа выражение принципа эквивалентности:
С(1/2Н2SО4).V(Н2SО4) =С(NаОН).V(NаОН).
В общем виде для любых случаев титрования это соотношение имеет следующее выражение:
Сн(Х).V(Х)=Сн(Т).V(Т),
аналитический катион химия реакция буферный
где Сн(Х), Сн(Т) - нормальности раствора анализируемого вещества Х и титранта Т, V(Х) и V(Т) - объемы анализируемого вещества и титранта соответственно. Из этого соотношения следует: V(Т)/V(Х)= Сн(Х)/Сн(Т).
Если известен объем анализируемого раствора, то по последнему уравнению можно рассчитать его нормальность: Сн(Х)= Сн(Т).V(Т)/V(Т).
Вычислив по результатам титрования нормальность анализируемого раствора из вышеприведенных уравнений можно определить массу вещества в любом объеме раствора.
МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСОНОМЕТРИИ ТЕМА 10
Индикаторы. Для фиксирования конца титрования используют визуальные (титрование с индикатором, цветным или флуоресцентным) и инструментальные методы (потснциометрическое, амперометрическое, фотометрическое титрование). Цветные индикаторы в кислотно-основном титровании - это слабые органические кислоты и основания, протонированные и непротонированные формы которых различаются по структуре и окраске. Существуют одноцветные (например, фенолфталеин) и двухцветные (например, метиловый оранжевый) индикаторы. Изменение окраски индикатора связано с таутомерией органических молекул, содержащих хромофор. Такие соединения обладают подвижными
л-электронами, и в зависимости от распределения электронной плотности молекуле можно приписать несколько структур; предельные структуры называются таутомерами. На распределение электронной плотности влияет наличие ауксохромных групп (например, NH^, ОНи т. п.). Ауксохромы связаны с ненасыщенным углеродным скелетом хромофора так, что положение двойных связей изменяется. Разность энергий основного и возбужденного (под действием света) состояний таких веществ мала, поэтому молекула поглощает свет в видимой части спектра, и вещество имеет определенную окраску. Например, метиловый оранжевый в щелочной среде окрашен в желтый цвет (хромофор -N=N-). В кислой среде азот, содержащий неподелённую пару электронов, протонируется, при этом образуется сопряженная система таутомеров
Цепь сопряжения в таутомере II, а следовательно, и подвижность П-электронов увеличивается, в результате энергия возбуждения молекул уменьшается и свет поглощается в красной области спектра. Метиловый оранжевый - двухцветный индикатор. Он относится к классу азоиндикаторов, имеющих в кислой среде красную, а в щелочной среде желтую окраску.
Метиловый красный
Сюда относятся также метиловый красный
Однако существуют соединения, не содержащие хромофоров, у которых под влиянием среды структура изменяется так, что появляются хромофорные группы. Таковы фталеины. Например, фенолфталеин в кислой среде бесцветен. В щелочной среде в результате перераспределения электронной плотности в его молекуле образуется хиноидная структура (хромофор), находящаяся в равновесии со своей таутомерной формой. Вещество приобретает красную окраску.
Группу индикаторов, схожую с фталеинами, составляют сульфофталеины: бромкрезоловый зеленый, феноловый красный, тимоловый синий и другие соединения. Например, в растворе фенолового красного в равновесии находятся красная и желтая формы.
Отдельную группу составляют трифенилметановые красители; кристаллический фиолетовый, метиловый фиолетовый, малахитовый зеленый и др.
Схематически (опуская промежуточные формы) равновесия в растворе индикатора можно представить как кислотно-основную реакцию
HIn + H2O£lrf+H3O+
Из константы кислотности индикатора, пренебрегая влиянием ионной силы и побочными взаимодействиями, получаем
Человеческий глаз воспринимает окраску одной из форм при определенной концентрации ее в растворе. Для двухцветного индикатора присутствие одной из форм заметно, если концентрация ее в несколько раз превышает концентрацию другой формы.
Очевидно, что, если глаз улавливает окраску одной из форм на фоне другой при большем или меньшем соотношении, а рН будет другим. Индикатор тем ценнее, чем меньше интервал рН.
Для одноцветных индикаторов при изменении рН нарастает или уменьшается концентрация их окрашенной формы. При определенной концентрации глаз замечает появление или исчезновение окраски. Пусть окрашенная форма In. Тогда рН, при котором появляется окраска, можно выразить.
Концентрация индикатора в растворе
Как видно, рН появления окраски зависит от концентрации индикатора в растворе. К индикаторам предъявляют ряд требований.
- . Индикатор должен обладать высоким светопоглощением, чтобы окраска даже его небольшого количества была заметна для глаза. Большая концентрация индикатора может привести к расходу на него титранта.
- . Переход окраски должен быть контрастным.
- . Область перехода окраски должна быть как можно уже. Сегзедида_области_перехода окраски индикатора (при этом рН = рКа) называется (pT) и фактически отождествляется с конечной точкой титрования.
Выбирают индикатор для титрования так, чтобы область перехода входила в скачок титрования. Границы скачка титрования определяются заданной точностью Чем жестче требования к точности титрования, тем уже скачок, тем более ограничен выбор индикатора.
Для сужения области перехода окраски и увеличения контрастности применяют смешанные индикаторы, которые составляют из индикатора и красителя. При определенном значении рН цвет красителя является дополнительным к цвету индикатора - в результате в этой точке окраска будет серой, а переход от окрашенного раствора к серому - контрастным.
На область перехода окраски индикатора (положение и интервал) влияют все факторы, от которых зависит константа равновесия (ионная сила, температура, посторонние вещества, растворитель), а также концентрация индикатора.
Для сульфофталеинов эта погрешность велика, для азоиндикаторов - мала. При небольших значениях ионной силы цвиттер-ионы, которые образует молекула в кислой среде (например, метиловый оранжевый), ведут себя как нейтральные молекулы.
Интервал перехода окраски индикатора смещается при нагревании: незначительно - у сульфофталеинов и фталеинов (индикаторов-кислот), существенно - у азоиндикаторов и других индикаторов-оснований. Это связано с увеличением Kw. Область перехода окраски метилового оранжевого, тропеолина и других смещается в кислую область: например у метилового оранжевого от 3,1-4,4 при 20 °С до 2,5-3,7 при 100 °С.
Влияние углекислого газа и веществ, образующих коллоидные системы.
За счет углекислого газа рН водного раствора уменьшается, поэтому все индикаторы с рТ > 4 чувствительны к СОг- На свойства индикаторов сильно влияет присутствие веществ, состоящих из макромолекул, например белков, образующих коллоидные системы. Во-первых наблюдается взаимодействие кислотных и основных групп белков и индикаторов, во-вторых, индикаторы адсорбируются на поверхности коллоидных частиц (индикаторы-основания - на отрицательно заряженных и индикаторы-кислоты - на положительно заряженных).
Специфические индикаторы - это вещества, которые образуют интенсивно окрашенное соединение" с одним из компонентов окислительно-восстановительной.
КТТ применяют индикаторы, т.н. металлохромные индикаторы - органические красители, имеющие свои хромофорные группы, способные обратимо изменять окраску при образовании комплексов с катионами металлов. Комплексонометрически контролируют жесткость воды. Общую жесткость воды (т. е. суммарное содержание в ней катионов магния и кальция, в мг-экв/л) определяют прямым титрованием стандартным р-ром ЭДТА в присутствии индикатора кислотного хромового темно-синего. Метод позволяет раздельно определять катионы металлов при их совместном присутствии, как в методе определения жесткости воды. Т. о. в аналитической химии широко применяются титриметрические методы определения различных в-в и элементов, причем в основе этих методов лежат правила качественного анализа и общие химические законы и свойства химических элементов и соединений. Методы окислительно-восстановительного титрования основаны на применении реакций, связанных с изменением степени окисления реагирующих веществ, т. е. на реакциях окисления - восстановления.
Разработано очень много методов окислительно-восстановительного титрования, их классифицируют в соответствии с применяемым стандартным раствором (титрантом).
Наиболее широкое применение получили методы перманганатометрии, хроматометрии, иодометрии.
Перманганатометрия.
В перманганатометрии стандартным раствором является раствор перманганата калия КмпО4. Сущность метода заключается в окислении восстановителей раствором перманганата калия. Окисление перманганатом калия можно проводить в кислой, нейтральной и в щелочной среде.
Следовательно, в данной реакции число эквивалентности КМпО4 равно 5. Молярная масса эквивалентов равна 1/5 его молярной массы: М (1/5КМпО4)=М(КМпО4):5= 158,04:5=31,61 г/моль.
Число эквивалентности FeSO4 равно 1. Молярная масса эквивалентов сульфата железа (II) равна его молярной массе: М (1/1 FeSO4) = M (FeSO4) = 151,91 г/моль.
Из электронных уравнений видно, что Мп присоединяет 3 электрона, а FeZT - 2. Следовательно, число эквивалентности z (KMnO4) = 3, FeSO4- 1. Молярные массы эквивалентов соответственно составят; М (1/3 КМпО4) = М (КМпО4): 3 = 52,68 г/моль; M(l/1 FeSO4) = M(FeSO4) = 151,91 г/моль.
Окисление перманганатом калия в щелочной среде. Например:
FeSO4+ KMnO4 +3KOH= Fe (OH)3+ K2 MnO4 + K2SO4
Согласно электронному уравнению молярные массы эквивалентов FeSO4 и КМпО4 равны их молярным массам, так как отдано и присоединено молекулами восстановителя и окислителя по одному электрону: z (FeSO4) = z (KMnO4)=l.
Иодометрия,
Иодометрия - метод титриметрического анализа, основанный на определении количества иода, которое затрачивается на окисление восстановителей или выделяется при взаимодействии окислителя с раствором иодида калия. Основная реакция метода описывается следующим уравнением:
2 + 2е~ <-* 2Г.
Эта реакция обратима. В зависимости от условий она может протекать в прямом или обратном направлениях. Иод представляет собой окислитель средней силы: стандартный потенциал системы равен 0,54 В. Поэтому сильные восстановители легко окисляются свободным иодом. Примером может быть окисление иодом ионов олова (II):
Sn2+ + I2=Sn4+ + 2Г.
Другие неорганические ионы в низших степенях окисления также можно определять посредством титрования раствором иода, например мышьяк(Ш), ванадий (IV), ртуть (Г). Таким способом определяют кислоты-восстановители, например сероводород, сернистую кислоту, тиосульфат и др. Иод окисляет также многие органические вещества: соединения, содержащие альдегидные группы, азот-, и серосодержащие соединения, оксисоединения и
Сильные окислители способны, наоборот, выделять иод из растворов иодидов. По этому типу происходит взаимодействие между иодидом калия и бихроматом или перманганатом калия, солями церия(1У), ванадия(У), железа(Ш), меди(П), органическими пероксидами и многими другими окислителями. Так, реакция между иодидом калия и ионами железа (III)
Fe3+ + 2Г = 2Fe2+ + 12
является основой иодометрического определения железа. Таким образом, эту реакцию можно использовать для определения и восстановителей, и окислителей. В первом случае применяют для титрования рабочий раствор иода, во втором - рабочий раствор тиосульфата натрия; последний реагирует с иодом в соответствии с уравнением
Na2S2O3 + h = Na2S4O6 + 2NaI.
Независимость двух основных реакций метода от кислотности раствора представляет важную особенность иодометрических определений. Окислительно-восстановительный потенциал системы 12/2Г не зависит от рН раствора, поэтому иодометрические определения можно проводить в широком интервале кислотности раствора (рН = 2-10). Это дает возможность подбирать в каждом отдельном случае самые благоприятные для определения окислителей или восстановителей условия кислотности и применять иодометрический метод для определения значительно более широкого круга объектов, чем в других методах титриметрического анализа. Иллюстрацией может быть определение мышьяка. Реакция окисления мышьяка (III) до мышьяка (V) проходит по уравнению:
HAsO2 + 2Н2О <-► H3AsO4 + 2Н+ + 2е\
Из уравнения видно, что во время реакции выделяются ионы водорода, следовательно, мышьяк окисляется легче в щелочном или в слабо щелочном растворе. Применять для титрования мышьяка такие окислители, как перманганат или бихромат калия, в этом случае неудобно. Перманганат восстанавливается до марганца (II) легче в сильно кислой среде; в слабо щелочном растворе окислительный потенциал перманганата снижается, вследствие чего реакция между перманганатом и мышьяком происходит очень медленно и не стехиометрически. Продуктами реакции являются соединения марганца (II) и марганца (IV), соотношение между количествами которых обычно не постоянно и зависит от условий определения. В то же время иодометрическое определение мышьяка проходит без всяких затруднений. Окислительный потенциал системы 12/2Г не изменяется под влиянием кислотности; рН раствора можно устанавливать только в зависимости от специфических условий протекания реакции.
Эта особенность иодометрического метода обусловливает его широкое применение при определении самых разнообразных неорганических и органических соединений.
Другая особенность иодометрии заключается в высокой точности установления точки эквивалентности, что связано с наличием чувствительного специфического индикатора. В качестве индикатора применяют раствор крахмала, который образует с наименьшими количествами иода окрашенное в интенсивно синий цвет адсорбционное соединение. Реакция отличается высокой чувствительностью - уже 0,00001 М растворы иода окрашиваются в присутствии крахмала в синий цвет. Точку эквивалентности можно также установить по желтой окраске свободного иода; повысить чувствительность в этом случае можно путем экстракции иода в слой органического растворителя (хлороформа, тетрахлорида углерода и др.). Таким способом можно перевести наименьшие количества иода из большого объема водного раствора в небольшой объем органического растворителя и значительно усилить интенсивность желтого окрашивания.
Существует две основных причины погрешностей при иодометрических определениях. Первая обусловлена летучестью иода и его растворов. Поэтому все работы, связанные с выделением иода, целесообразно проводить в закрытых склянках или колбах. Вследствие летучести иода и его малой растворимости в воде для титрования применяют растворы иода в концентрированном растворе иодида калия. При определении окислителей создают в растворе большой избыток иодида калия, чтобы перевести выделяющийся при реакции твердый иод в раствор и уменьшить его летучесть. Вторая причина погрешностей - окисление растворов иодида калия кислородом воздуха.
Броматометрия.
Бромат калия в кислом растворе проявляет достаточно сильные окислительные свойства. Электронно-ионную реакцию восстановления бромат-ионов можно представить следующим уравнением:
ВЮз' + 6YT + бе" = Вг" + ЗН2О, Ео = 1,46 В.
Однако в кислом растворе равновесие между ионами бромата и бромида невозможно, так как они взаимодействуют между собой с образованием свободного брома:
ВгОз'+бН*" + 5Вг" = ЗВг2 + ЗН2О.
Таким образом, окислителем по существу является свободный бром, потенциал которого значительно ниже, чем потенциал бромата:
Вг" = 2е" + Вг2, Е0=1,07В.
Эквивалент бромата калия, как это видно из уравнений, равен одной шестой части молекулярной массы.
Бромат калия легко очистить от примесей перекристаллизацией из водного раствора. Перекристаллизованную соль высушивают в сушильном шкафу при 100 - 150°С и сохраняют в закрытой склянке. Рабочий раствор КВгО3 можно готовить непосредственно из навески высушенной соли. Иногда к рабочему раствору прибавляют при его приготовлении избыток бромида калия; в этом случае при титровании таким бромид-броматным раствором в кислой среде сразу выделяется свободный бром, который реагирует с находящимся в растворе восстановителем.
Рабочий раствор бромата калия обычно применяют в следующих случаях.
. Для прямого титрования неорганических ионов, находящихся в низшей степени окисления. Известны методы определения трехвалентного мышьяка, пероксида водорода, олова (II), меди(1); таллия (I), железа(П), урана(ГУ), ванадия(П) и др. Наиболее распространен броматометрический метод титрования сурьмы (III):
КВЮз + 3SbCl3 + 6HC1 = KBr + 3SbCl5 + ЗН2О.
Титрование обычно проводят в растворе хлороводородной кислоты.
2. Для прямого определения некоторых органических веществ. Примером может служить титрование аскорбиновой кислоты (витамина С) в различных медицинских препаратах.
Известны и другие примеры. Так, тиомочевина количественно окисляется броматом по реакции:
3CS(NH2)2 + 4НВгО3 + ЗН2О = 3CO(NH2)2 + 3H2SO4 + 4HBr.
Продуктом реакции является мочевина.
. Для определения некоторых ароматических органических соединений и косвенного определения многих неорганических ионов по реакциям замещения.
Фенолы и его производные титруются раствором бромид-бромата с образованием соответствующих бромзамещенны.х соединений. В случае фенола продуктом реакции является трибромфенол:
С6Н5ОН + ВгОз" + 5Br" + 6¥t = СбН2Вг30Н + ЗНВг + ЗН2О.
Здесь происходит реакция бромирования фенола. Аналогичные реакции бромирования лежат в основе определения многих других органических соединений ароматического ряда, например салициловой кислоты, крезолов, динитрофенолов, резорцина, Р-нафтола, анилина, антипирина и др.
Для косвенного определения металлов большое значение имеет реакция бромата калия с 8-гидроксихинолином. В кислой среде 8-гидроксихинолин реагирует с бромид-броматной смесью по уравнению:
8-Гидроксихинолин образует с многими металлами (алюминием, железом, титаном, медью, кадмием, цинком, кобальтом, никелем, магнием и др.) нерастворимые осадки гидроксихинолинатов. Селективность осаждения достигается регулированием рН раствора. Так, полное осаждение гидроксихинолинатов алюминия, железа, титана, меди, магния, свинца достигается при рН 4,4; 2,8; 4,8; 3,3; 8,2 и 8,4 соответственно. Поэтому можно, например, осаждать алюминий в присутствии магния, железо - в присутствии свинца и т. д. Другой путь повышения селективности - введение маскирующих веществ. Описанные приемы позволяют определять один металл в присутствии ряда других.
Хроматометрия.
Хроматометрия - метод титрования раствором бихромата калия. Электронно-ионная реакция восстановления K2G12O7 протекает в соответствии со следующим уравнением:
Сг2О72' + 14FT + бе' = 2Сг3+ + 7Н2О.
Стандартный потенциал этой системы Ео = 1,36 В.
Из уравнения видно, что эквивалент бихромата калия равен 1/6 молекулярной массы (фактор эквивалентности равен 1/6).
Стандартный потенциал бихромата калия ниже, чем у перманганата, но все же достаточно высок. Это позволяет титровать почти все те вещества, которые определяют методом перманганатометрии. Однако преимуществом К2Сг207 является то, что при титровании в растворе НС1 не наблюдается окисления хлорид-ионов до свободного хлора, как в перманганатометрии. Другое преимущество состоит в устойчивости растворов бихромата - концентрация рабочего раствора при длительном хранении в закрытых сосудах остается практически неизменной. Бихромат калия легко очистить перекристаллизацией; полученный препарат высушивают при 130 - 150 °С. Рабочий раствор нужной концентрации можно приготовить непосредственно из навески высушенного препарата.
Бихромат калия восстанавливается до хрома (III); ионы последнего окрашены в зеленый цвет. Поэтому для установления конечной точки титрования необходимо применять специальные индикаторы.
Самое большое распространение получил метод хроматометрического определения железа(П)
Сг2О72" + 6Fe2+ + 14Н+ = 2Cr3+ + 6Fe3+ + 7Н2О. (22.14)
Титровать можно в растворах хлороводородной кислоты, причем никаких побочных реакций не происходит.
Некоторым недостатком по сравнению с перманганатометрическим определением является необходимость использования специальных индикаторов. Обычно применяют обратимые окислительно-восстановительные индикаторы - дифениламин, фенилантраниловую кислоту и др.
Методы предварительного восстановления.
Методами окисления - восстановления можно определять любые окислители или восстановители. Описано применение для этой цели растворов перманганата, бихромата, бромата калия, иода. Используют и другие окислители, например Ce(SO4)2, NH4VO3. Известны и методы титрования окислителей восстановителями, например растворами FeSO-ь TiCl3 (титанометрия), SnCl2, CrCl2 (хромометрия). Эти методы распространены мало, так как рабочие растворы сильных восстановителей необходимо защищать от окисления кислородом воздуха, сохраняя их и проводя титрование в атмосфере инертного газа, что требует применения специальных приспособлений. Кроме того, для фиксации конечной точки титрования нужны окислительно-восстановительные индикаторы, что не всегда удобно или доступно.
При титровании некоторыми окислителями нельзя обходиться без индикаторов, например при использовании рабочих растворов сульфата церия, ванадата аммония, бихрсмата калия. Поэтому наибольшее распространение в практике получили методы титрования окислителями, в частности растворами перманганата и иода; в первом случае индикатором служит сам рабочий раствор, во втором - специфический по отношению к иоду индикатор - крахмал. Высокая устойчивость растворов бихромата калия и возможность непосредственного использования его в качестве исходного вещества нередко делают применение К2С12О7 более удобным по сравнению с другими окислителями. Кроме того, потенциал бихромата калия ниже, чем у перманганата, вследствие чего титрование первым проходит без побочных реакций, мешающих определению. Бромат калия лучше всего применять в качестве титрднта для определения ряда органических веществ, с которыми он реагирует стехиометрически с образованием различных бромзамещенных соединений.
Несмотря на перечисленные достоинства, применение окислителей связано со следующими недостатками. Обычно предварительная подготовка пробы к анализу состоит в переведении анализируемого материала в раствор посредством обработки различными кислотами; чаще всего применяют азотную кислоту или ее смесь с хлороводородной или серной кислотой. Так, медные сплавы растворяют в азотной кислоте, причем содержащиеся в них элементы - железо, олово и другие - превращаются в соединения высших степеней окисления. При анализе различных чугунов и сталей необходимо определять ванадий, молибден, вольфрам, титан и некоторые другие легирующие элементы, которые вследствие обработки пробы окислительными агентами также содержатся в полученном растворе в высших степенях окисления. Железные руды содержат оксиды железа; растворяя их в хлороводородной кислоте с добавками различных окислителей, получают железо в степени окисления +3 и т. д.
титрование окислителями - КМ11О4, K2C12O7 и другими - возможно только после переведения определяемых элементов в низшие степени окисления. Восстановление необходимо провести количественно, для чего обычно вводят больший или меньший избыток восстановителя. Перед титрованием этот избыток необходимо каким-либо способом устранить, иначе рабочий раствор окислителя будет расходоваться не только на реакцию с определяемым элементом, но также на окисление восстановителя, и результат титрования окажется неправильным.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЙ, физические свойства веществ, качественный и количественный состав ТЕМА 11
Указанные методы анализа применяются в случае присутствия зависимости между измеряемыми физическими свойствами веществ и их качественным и количественным составом. Поскольку для измерения физических св-в в-в применяются различные приборы (инструменты), то эти методы наз-ся инструментальными. Классификация физических и физико-химических методов анализа. Основана на учете измеряемых физических и физико-химических св-в в-ва или изучаемой системы. Оптические методы основаны на измерении оптических св-в в-в. Хроматографические на использовании способности различных в-в к избирательной сорбции.
Электрохимические методы основаны на измерении электрохимических св-в системы.
Радиометрические основаны на измерении радиоактивных св-в в-в. Термические на измерении тепловых эффектов соответствующих процессов. Масс-спектрометрические на изучении ионизированных фрагментов («осколков») в-в. Ультразвуковые, магнитохимические, пикнометрические и т.д.
Достоинства инструментальных методов анализа: низкий предел обнаружения 1 -10-9мкг; малая предельная концентрация, до 10-12г/мл определяемого в-ва; высокая чувствительность, формально определяемая величиной тангенса угла наклона соответствующей градуировочной кривой, отражающей графически зависимость измеряемого физического параметра, который откладывается обычно по оси ординат, от кол-ва или концентрации определяемого в-ва (ось абсцисс). Чем больше тангенс угла наклона кривой к оси абсцисс, тем чувствительнее метод, что означает следующее: для получения одинакового «отклика» - изменения физического свойства - требуется меньшее изменение концентрации или кол-ва измеряемого в-ва. К достоинствам относится высокая селективность (избирательность) методов, т. е. сотавные компоненты смесей можно определять без разделения и выделения этих компонентов; малая продолжительность времени проведения анализа, возможность их автоматизации и компьютеризации. Недостатки: сложность аппаратуры и высокая стоимость; большая погрешность (5 -20 %), чем в классическом химич-ом анализе (0,1 -0,5%); хуже воспроизводимость. Оптические методы анализа основаны на измерении оптических св-в в-ва (испускание, поглощение, рассеяние, отражение, преломление, поляризация света), проявляющихся при взаимодействии электромагнитного излучения с в-вом.
Классификация по изучаемым объектам: атомный и молекулярный спектральный анализ. По характеру взаимодействия электромагнитного излучения с в-ом. При этом различают следующие методы. Атомно-абсорбционный анализ, в основе которого лежит измерениепоглощения монохроматического излучения атомами определяемого в-ва в газовой фазе после атомизации в-ва. Эмиссионный спектральный анализ - измерение интенсивности света, излучаемого в-ом (чаще всего атомами или ионами) при его энергетическом возбуждении, например, в плазме электрического разряда. Пламенная фотометрия - использование газового пламени в качестве источника энергетического возбуждения излучения.
Нефелометрия - измерение рассеивания света частицами света дисперсной системы (среды).
Турбидиметрический анализ - измерение ослабления интенсивности излучения при его прохождении через дисперсную среду. Рефрактометрический анализ измерение показателей светопреломления в-в. Поляриметрический анализ измерение величины оптического вращения - угла вращения плоскости поляризации света оптически активными в-ми. По области используемого электромагнитного спектра классифицируют следующие методы: спектроскопия (спектрофотометрия) в УВИ области спектра, т. е. в ближайшей ультрафиолетовой области спектра - в интервале длин волн 200 - 400 нм и в видимой области - в интервале длин волн 400 - 700 нм. Инфракрасная спектроскопия, изучающая участок электромагнитного спектра в интервале 0,76 - 1000 мкм (1 мкм=10-6 м), реже рентгеновская и микроволновая спектроскопия. По природе энергетических переходов в различных спектрах - электронных (изменение энергии электронных состояний атомов, ионов, радикалов, молекул, кристаллов в УВИ области); колебательных (при изменении энергии колебательных состояний 2-х и многоатомных ионов, радикалов, молекул, а также жидких и твердых фаз в ИК области); вращательных также в ИК и микроволновой области. Т.о. взаимодействие между молекулами и электромагнитным излучением заключается в том, что путем поглощения электромагнитного излучения молекулы переходят в возбужденное состояние. При этом важную роль играет энергия, т. е. длина волны поглощенного излучения.
Так, в рентгеновских лучах, длина волны которых 0,05 - 5 нм, происходит процесс возбуждения внутренних электронов в атомах и молекулах; в ультрафиолетовых лучах (5 - 400 нм) происходит процесс возбуждения внешних электронов в атомах и молекулах; видимый свет (400 - 700 нм) происходит возбуждение внешних электронов в сопряженных π-электронных системах; инфракрасное излучение (700 нм - 500 мк) происходит процесс возбуждения колебаний молекул; микроволны (500 мк - 30 см) процесс возбуждения вращения молекул; радиоволны (более 30 см) процесс возбуждения спиновых переходов в атомных ядрах (ядерный магнитный резонанс).
Поглощение излучений позволяет в спектрометрии их измерять и регистрировать. При этом падающее излучение делится на эталонное и измеряемое при одинаковой интенсивности. Измеряемое излучение проходит через пробу; при этом происходит поглощение, изменяется интенсивность. При поглощении энергии электромагнитного излучения частицы в-ва (атомы, молекулы, ионы) увеличивают свою энергию, т. е. переходят в более высоколежащее энергетическое состояние. Электронные, колебательные, вращательные энергетические состояния частиц в-ва могут изменяться лишь дискретно, на строго определенную величину. Для каждой частицы существует индивидуальный набор энергетических состояний - энергетических уровней (термов), например, электронных уровней энергии. Электронные энергетические уровни молекул и многоатомных ионов имеют тонкую структуру - колебательные подуровни; поэтому одновременно с чисто электронными переходами осуществляются и колебательные переходы.
Полоса в электронном спектре поглощения
Каждому электронному (электронно-колебательному) переходу с нижнего энергетического уровня на более высоко лежащий электронный уровень отвечает полоса в электронном спектре поглощения.
Так как разность между электронными уровнями для каждой частицы (атома, иона, молекулы) строго определенна, то строго определенным является и положение полосы в электронном спектре поглощения, соответствующей тому или иному электронному переходу, т. е. длина волны (частота, волновое число) максимума полосы поглощения. Различия в интенсивности измеряются детектором и записываются на самописце в виде сигнала (пика), стр 318, химия, справочник школьника и студента, схема спектрометра. Ультрафиолетовая спектроскопия и абсорбционная спектроскопия в видимой области.
Переходы электронов в молекулах с занятых на незанятые энергетические уровни
Поглощение электромагнитного излучения из ультрафиолетовой и видимой части спектра; возбуждает переходы электронов в молекулах с занятых на незанятые энергетические уровни. Чем больше разность в энергии между энергетическими уровнями, тем большую энергию, т.е. более короткую длину волны, должно иметь излучение. Часть молекулы, которая в значительной части определяет поглощение света, называется хромофором (буквально, несущие цвет) - это атомные группы, влияющие на поглощение света молекулой, в особенности сопряженные и ароматические системы π-электронов.
Структурные элементы хромофоров в основном и участвуют в поглощении кванта световой энергии, что приводит к появлению полос в сравнительно узком участке спектра поглощения соединений. Практическое значение для определения строения органических молекул имеет область от 200 до 700 нм. Количественное измерение: наряду с положением максимума поглощения для анализа важно значение экстинкции (ослабления) излучения, т. е. интенсивности его поглощения.
В соответствии с законом Ламберта - Бера Е=lgI0 /I=εcd, Е - экстинкция, I0 - интенсивность падающего света, I - интенсивность проходящего света, ε - молярный коэффициент экстинкции, см2/моль, c - концентрация, моль/л, d - толщина слоя пробы, см.
Экстинкция зависит от концентрации поглощающего в-ва. Методы абсорбционного анализа колориметрия, фотоэлектроколориметрия, спектрометрия.
Колориметрия метод анализа на визуальном сравнении окраски жидкостей определение рН
Колориметрия самый простой и старый метод анализа, основан на визуальном сравнении окраски жидкостей (определение рН почвы на приборе Алямовского) - самый простой метод сравнения с серией эталонных р-ов. Широко распространены 3-и метода колориметрии: метод стандартных серий (метод шкалы), метод уравнивания окрасок и метод разбавления. Используются стеклянные колориметрические пробирки, стеклянные бюретки, колориметры, фотометры. М
етод шкалы - это определение рН на приборе Алямовского, т. е. серия пробирок с различной конц-ей в-ва и разная по изменению интенсивности цвета р-ра или эталонных р-ов. Фотоколориметрия - метод основан на измерении интенсивности немонохроматического светового потока, прошедшего через анализируемый р-р с помощью фотоэлементов.
Световой поток от источника излучения (лампы накаливания) проходит через светофильтр, пропускающий излучение лишь в определенном интервале длин волн, через кювету с анализируемым р-ом и попадает на фотоэлемент, преобразующий световую энергию в фототок, регистрируемый соответствующим прибором. Чем больше светопоглощение анализируемого р-ра (т. е. чем выше его оптическая плотность), тем меньше энергия светового потока, попадающего на фотоэлемент. ФЭКи снабжаются н-ми светофильтрами, имеющими максимум светопропускания при различных длинах волн. При наличии 2-х фотоэлементов происходит измерение 2-х световых потоков, одного через анализируемый р-р, другого через р-р сравнения. Концентрацию исследуемого в-ва находят по градуировочному графику.
Электрохимические методы анализа основаны на электродных реакциях и на переносе электричества через р-ры.
В количественном анализе используется зависимость величин измеряемых параметров электрохимических процессов (разность электрических потенциалов, ток, кол-во электричества) от сод-ия определяемого в-ва в р-ре, участвующего в данном электрохимическом процессе. Электрохимические процессы - такие процессы, которые сопровождаются одновременным протеканием химических реакций и изменением электрических св-в системы, которую в подобных случаях можно наз-ать электрохимической системой. Основные принципы потенциометрии
Как следует из названия метода - в нем измеряется потенциал. Для пояснения, что за потенциал и почему он возникает, рассмотрим систему состоящую из металлической пластины и находящегося с ней в контакте раствора, содержащего ионы того же металла (электролит) (рис. 1).
Электрод - система
Данная система называется электродом. Любая система стремится к такому состоянию, которое отвечает минимуму ее внутренней энергии. Поэтому в первый момент после погружения металла в раствор на границе раздела фаз начинают протекать процессы, ведущие к снижению внутренней энергии системы. Предположим, что ионизированное состояние атома металла энергетически более «выгодно», чем нейтральное (возможен и обратный вариант). Тогда в первый момент времени атомы металла будут переходить из поверхностного слоя пластины в раствор, оставляя в ней свои валентные электроны.
При этом поверхность пластины приобретает отрицательный заряд, причем этот заряд растет по мере увеличения количества атомов металла, перешедших в виде ионов в раствор. Электростатические силы притяжения разноименных зарядов (отрицательнозаряженные электроны в пластине и положительные ионы металла в растворе) не позволяют удалиться этим зарядам от границы раздела фаз, а также вызывают обратный процесс перехода ионов металла из раствора в металлическую фазу и восстановления их там. Когда скорости прямого и обратного процессов становятся одинаковыми, наступает равновесие. Состояние равновесия системы характеризуется разделением зарядов на границе раздела фаз, т. е. появляется «скачок» потенциала. Следует отметить, что описанный механизм возникновения электродного потенциала является не единственным, в реальных системах протекает также множество других процессов, приводящих к образованию «скачка» потенциалов на межфазовой границе. Кроме того, «скачок» потенциала может возникать на границе раздела фаз не только при контакте электролита с металлом, но и при контакте электролита с другими материалами, например, полупроводниками, ионообменными смолами, стеклами и т. д.
При этом ионы, концентрация которых влияет на потенциал электрода называются потенциалопределяющими. Потенциал электрода зависит от природы материала, контактирующего с электролитом, концентрации потенциалопределяющих ионов в растворе и температуры. Этот потенциал измеряется относительно другого электрода, потенциал которого постоянен. Т. о., установив эту связь, возможно использовать ее в аналитической практике для определения концентрации ионов в растворе. При этом электрод, потенциал которого измеряется, носит название измерительный, а электрод, относительно которого производятся измерения - вспомогательный или электрод сравнения.
Постоянство потенциала электродов сравнения достигается постоянством концентрации потенциалопределяющих ионов в его электролите (электролит №1). Состав электролита №2 может меняться. Для предотвращения смешивания двух разных электролитов они разделяются мембраной, проницаемой для ионов. Потенциал измерительного электрода принимается равным измеренной э.д.с., приведенной электрохимической системы. Применяя в качестве электролита №2, растворы известного состава можно установить зависимость потенциала измерительного электрода от концентрации потенциалоопределяющих ионов. Эта зависимость в дальнейшем может быть использована при анализе раствора неизвестной концентрации.
Для стандартизации шкалы потенциалов в качестве электрода сравнения принят стандартный водородный электрод, потенциал которого принят равным нулю при любой температуре. Однако при обычных измерениях водородный электрод применяется редко из-за своей громоздскости. В повседневной практике применяют другие более простые электроды сравнения, потенциал которых относительно водородного электрода определен. Поэтому, при необходимости, результат измерения потенциала, проведенного относительно таких электродов, может быть пересчитан относительно водородного электрода. Наиболее широко распространенными являются хлорсеребряный и каломельный электроды сравнения. Разность потенциалов измерительного электрода и электрода сравнения является мерой концентрации определяемых ионов.
Электродную функцию можно описать с помощью линейного уравнения Нернста:
Е = Е0 + 2,3 RT/nF *lg а,
где Е - разность потенциалов между измерительным электродом и электродом сравнения, мВ; Е0 - константа, зависящая в основном от свойств электрода сравнения (стандартный потенциал электрода), мВ; R - газовая постоянная, Дж*моль-1* К-1.; n - заряд иона с учетом его знака; F - число Фарадея, Кл/моль; Т - абсолютная температура, 0 К; член 2,3 RT/nF, входящий в уравнение Нернста при 250 С равен 59,16 мВ для однозарядных ионов. Метод без наложения внешнего (постороннего) потенциала классифицируется как метод, основанный на учете природы источника электрической энергии в системе. В этом методе источником эл.эн. служит сама элек-хим-ая система, представляющая собой гальванический элемент (гальваническую цепь) - потенциометрические методы. ЭДС и электродные потенциалы в такой системе зависят от сод-ия определяемого в-ва в р-ре. Электрохимическая ячейка включает 2-ва электрода - индикаторный и электрод сравнения. Величина ЭДС, генерируемой в ячейке, равна разности потенциалов этих 2-х электродов.
Потенциал электрода сравнения в условиях проведения потенциометрического определения остается постоянным, то ЭДС зависит только от потенциала индикаторного электрода, т. е. от активностей (концентраций) тех или иных ионов в р-ре. На этом и основано потенциометрическое определение концентрации данного в-ва в анал-ом р-ре. Применяют как прямую потенциометрию, так и метод потенциометрического титрования.
При определении рН р-ов в кач-ве индикаторных используются электроды потенциал которых зависит от конц-ии ионов водорода: стеклянный, водородный, хингидронный (окислительно-восстановительный электрод в виде платиновой проволоки, погруженной в р-р НС1, насыщенной хингидроном - эквимолекулярным соединением хинона с гидрохиноном) и нек-ые др. Мембранные или ион-селективные электроды имеют реальный потенциал, зависящий от активности тех ионов в р-ре, кот-ые сорбируются мембраной электрода (твердой или жидкой) метод наз-ся ионометрией.
Спектрофотометрами наз-ют приборы, позволяющие производить измерения светопоглощения образцов в узких по спектральному составу пучках света (монохроматический свет). Спектрофотметры позволяют разлагать белый свет в непрерывный спектр, выделять из этого спектра узкий интервал длин волн (1 - 20 нм ширина выделяемой полосы спектра), пропускать изолированный пучок света через анализируемый р-р и измерять с высокой точностью интенсивность этого пучка. Поглощение света окрашенным в-ом в р-ре измеряют, сравнивая его с поглощением нулевого р-ра. В спектрофотометре сочетаются два прибора: монохроматор для получения монохроматического светового потока и фотоэлектрический фотометр, предназначенный для измерения интенсивности света. Монохроматор состоит из источника света, диспергирующего устройства (разлагающего белый свет в спектр) и устройства регулирующего величину интервала длин волн светового пучка, падающего на р-р.
Из разнообразных физико-химических и физических методов анализа наибольшее значение имеют 2-ве группы методов:
- 1 - методы, основанные на изучении спектральных характеристик в-ва;
- 2 - методы, основанные на изучении физико-химических параметров.
Спектральные методы основаны на явлениях, происходящих при взаимодействии вещества с различными видами энергии (электромагнитным излучением, термической энергией, электрической и пр.). К основным видам взаимодействия в-ва с лучистой энергией относится поглощение и испускание (эмиссия) излучения. Характер явлений, обусловленных поглощением или испусканием, в принципе одинаков. При взаимодействии излучения с в-вом частицы его (атомы молекулы) переходят в возбужденное состояние. Через некоторое время (10-8 с) частицы возвращаются в основное состояние, испуская избыточную энергию в виде электромагнитного излучения. Эти процессы связаны с электронными переходами в атоме или молекуле.
Электромагнитное излучение можно охарактеризовать длиной волныλ или частотой ν, которые связаны между собой соотношением ν=с/λ, где с - скорость света в вакууме (2,298108 м/с). Совокупность всех длин волн (частот) электромагнитного излучения составляет электромагнитный спектр от γ-лучей (коротковолновая область, фотоны обладают высокой энергией) до видимой области спектра (400 - 700 нм) и радиоволн (длинноволновая область, фотоны с низкой энергией).
На практике имеют дело с излучением, характеризующимся определенным интервалом длин волн (частот), т. е. с определенным участком спектра (или, как говорят, с полосой излучения). Часто для аналитических целей используется и монохроматический свет (световой поток, в котором электромагнитные волны имеют одну длину волны). Избирательное поглощение атомами и молекулами излучения с определенными длинами волн приводит к тому, что каждое в-во характеризуется индивидуальными спектральными характеристиками.
Для аналитических целей используют как поглощение излучения атомами и молекулами (соответственно атомно- абсорбционная спектроскопия), так и испускание излучения атомами и молекулами (эмиссионная спектроскопия и люминесценция).
Спектрофотометрия основана на избирательном поглощении электромагнитного излучения в-вом. Измеряя поглощение в-вом излучения различных длин волн, можно получить спектр поглощения, т. е. зависимость поглощения от длины волны падающего света. Спектр поглощения - это качественная характеристика в-ва. Количественной характеристикой является количество поглощенной энергии или оптическая плотность раствора, которая зависит от концентрации поглощающего в-ва по закону Бугера-Ламберта-Бера: D=εІс, где D - оптическая плотность, i - толщина слоя; с - концентрация, моль/л; ε - молярный коэффициент поглощения (ε = D при І=1 см и с=1 моль/л). Величина ε служит характеристикой чувствительности: чем больше значение ε, тем меньшие количества в-ва можно определить.
Многие в-ва (особенно органические) интенсивно поглощают излучение в УФ- и видимой областях, что делает возможным их непосредственное определение. Большинство ионов, наоборот, слабо поглощают излучение видимой области спектра (ε ≈ 10…1000), поэтому их обычно переводят в другие, более интенсивно поглощающие соединения, а затем проводят измерения. Для измерения поглощения (оптической плотности) используют спектральные приборы 2-х видов: фотоэлектроколориметры (с грубой монохроматизацией) и спектрофотометры (с более тонкой монохроматизацией). Наиболее распространенным является фотометрический метод анализа, количественные определения в котором основаны на законе Бугера-Ламберта-Бера. Основными приемами фотометрических измерений являются: метод молярного коэффициента светопоглощения, метод градуировочного графика, метод стандартов (метод сравнения), метод добавок. В методе молярного коэффициента светопоглощения измеряют оптическую плотность D исследуемого р-ра и по известному значению молярного коэффициента светопоглощения ε рассчитывают концентрацию с поглощающего в-ва в растворе: с = D/(ε І). В методе градуировочного графика готовят ряд стандартных растворов с известным значением концентрации с определяемого компонента и определяют их значение оптической плотности D.
По полученным данным строят градуировочный график - зависимость оптической плотности раствора от концентрации в-ва: D = f(с). В соответствии с законом Бухера-Ламберта-Бера график представляет собой прямую линию. Затем измеряют оптическую плотность D исследуемого раствора и по градуировочному графику определяют концентрацию определяемого соединения.
Метод сравнения (стандартов) основан на сравнении оптической плотности стандартного и исследуемого растворов:
ст=ε*І*сст и Dх= ε*І*сх,
откуда Dх/ Dст=ε*І*сх/ε*І*сст и сх=сст*Dх/Dст. В методе добавок сравниваются значения оптической плотности исследуемого раствора и того же раствора с добавлением (са) известного количества определяемого компонента.
По результатам определений рассчитывают концентрацию в-ва в исследуемом растворе:
Dх= ε*І*сх и Dх+а= ε*І*(сх+са), откуда Dх/Dх+а= ε*І*сх/ε*І*(сх +са) и сх=са* Dх/Dх+а - Dх..
Атомно-абсорбционная спектроскопия основана на избирательном поглощении излучения атомами. Для переведения вещества в атомарное состояние раствор образца впрыскивают в пламя или подогревают в специальной кювете. В результате растворитель улетучивается или сгорает, а твердое в-во атомизируется. Большая часть атомов остается в невозбужденном состоянии, и лишь небольшая часть возбуждается с последующим испусканием излучения. Набор линий, соответствующий длинам волн поглощаемого излучения, т. е. спектр, является качественной характеристикой, а интенсивность этих линий - соответственно количественной характеристикой в-ва.
Атомно-эмиссионная спектроскопия основана на измерении интенсивности света, излучаемого возбужденными атомами. Источниками возбуждения могут быть пламя, искровый разряд, электрическая дуга и др. Для получения спектров испускания пробу в виде порошка или раствора вводят в источник возбуждения, где происходит переход в-ва в газообразное состояние или частичный распад его на атомы и простые (по составу) молекулы. Качественной характеристикой в-ва является его спектр (т. е. набор линий в спектре испускания), а количественной - интенсивность этих линий.
Люминесценция основана на испускании излучения возбужденными молекулами (атомами, ионами) при переходе их в основное состояние. Источниками возбуждения при этом могут быть ультрафиолетовое и видимое излучение, катодные лучи, энергия химической реакции и пр. Энергия излучения (люминесценции) всегда меньше поглощенной энергии, т. к. часть поглощенной энергии еще до начала испускания преобразуется в тепловую.
Следовательно, люминесцентное испускание всегда имеет меньшую длину волны, чем длина волны поглощенного при возбуждении света. Люминесценция может использоваться как для обнаружения в-в (по длине волны), так и для их количественного определения (по интенсивности излучения). Электрохимические методы анализа основаны на взаимодействии в-ва с электрическим током. Протекающие при этом процессы локализованы либо на электродах, либо в приэлектродном пространстве. Большинство методов относятся к первому из этих типов.
Потенциометрия.
Электродным процессом называется гетерогенная реакция, при которой заряженная частица (ион, электрон) переносится через границу раздела фаз. В рез-те такого переноса на пов-ти электрода возникает разность потенциалов, обусловленная образованием двойного электрического слоя. Как всякий процесс, электродная реакция с течением времени приходит к равновесию, и на электроде устанавливается равновесный потенциал.
Измерение величин равновесных электродных потенциалов является задачей потенциометрического метода анализа. Измерения при этом проводят в электрохимической ячейке состоящей из 2-х полуэлементов. Одиг из них содержит индикаторный электрод (потенциал которого зависит от концентрации определяемых ионов в растворе в соответствии с уравнением Нернста), а другой - электрод сравнения (потенциал которого постоянен и не зависит от состава раствора). Метод может быть реализован в варианте прямой потенциометрии или в варианте потенциометрического титрования.
В первом случае измеряют потенциал индикаторного электрода в анализируемом растворе относительно электрода сравнения и по уравнению Нернста рассчитывают концентрацию определяемого иона. В варианте потенциометрического титрования определяемый ион титруют подходящим реагентом, следя одновременно за изменением потенциала индикаторного электрода.
По полученным данным строят кривую титрования (зависимость потенциала индикаторного электрода от объема прибавленного титранта). На кривой вблизи точки эквивалентности наблюдается резкое изменение значения потенциала (скачок потенциала) индикаторного электрода, что позволяет рассчитать содержание определяемого иона в растворе. Электродные процессы очень многообразны.
В целом их можно классифицировать на 2-ве большие группы:
процессы, происходящие с переносом электронов (т. е. собственно электрохимические процессы), и процессы, связанные с переносом ионов( при этом электроду присуща ионная проводимость). В последнем случае речь идет о так называемых ионселективных мембранных электродах, широко применяемых в настоящее время. Потенциал такого электрода в растворе, содержащем определяемые ионы, зависит от их концентрации по уравнению Нернста. К этому же типу электродов относится и стеклянный электрод, применяемый в рН-метрии.
Возможность создания большого числа мембранных электродов с высокой селективностью к тем или иным ионам выделила эту область потенциометрического анализа в самостоятельную отрасль - ионометрию.
Полярография.
При прохождении тока в электрохимической ячейке наблюдается отклонение величин электродных потенциалов от их равновесных значений. В силу ряда причин возникает так называемая электродная поляризация. Явление поляризации, возникающей в процессе электролиза на электроде с малой поверхностью, лежит в основе данного метода анализа. В этом методе к электродам, опущенным в исследуемый раствор, прикладывают возрастающую разность потенциалов. При малой величине разности потенциалов ток через раствор практически не идет (т. н. остаточный ток). При увеличении разности потенциалов до величины, достаточной для разложения электролита, сила тока резко возрастает.
Потенциал разложения, величина
Данную величину разности потенциалов называют потенциалом разложения. Измеряя зависимость силы тока, проходящего через раствор, от величины приложенного напряжения, можно построить т. н. вольтамперную кривую, которая позволяет с достаточной точностью определить качественный и количественный состав раствора.
При этом качественной характеристикой в-ва является величина разности потенциалов, достаточная для его электрохимического разложения (потенциал полуволны ЕS), а количественной - величина прироста силы тока, обусловленная его электрохимическим разложением в растворе (высота длины волны Н, или различие в величинах предельного диффузионного тока и остаточного тока). Для количественного определения концентрации в-ва в растворе используют следующие приемы: метод градуировочного графика, метод стандартов, метод добавок. Кондуктометрический метод анализа основан на зависимости электропроводности раствора от концентрации электролита. Применяется, как правило, в варианте кондуктометрического титрования, точку эквивалентности в котором определяют по перегибу кривой титрования (зависимости электропроводности от количества прибавленного титранта).
Читать больше..Амперометрическое титрование является разновидностью потенциометрического титрования, только индикаторным электродом является полярографическое устройство, т.е. применяется микроэлектрод с наложенным напряжением.
метод определения различных физико-химических величин
- 17 января 2019
- потенциометрия электрод индикаторные электроды титрование рh раствор ионометрия электрод сравнения pH-электрод pH электрод pH-метра водородный показатель ионы водорода измерение ph
метод определения различных физико-химических величин
Читать больше..Процедура, потенциометрия
- 17 января 2019
- потенциометрия электрод индикаторные электроды титрование рh раствор ионометрия электрод сравнения pH-электрод pH электрод pH-метра водородный показатель ионы водорода измерение ph
Процедура, потенциометрия В настоящее время легко доступны потенциометрические индикаторные электроды, такие как стеклянные электроды для измерения pH и окислительно-восстановительные электроды. С помощью этих электродов pH или окислительно...
Читать больше..